Scientific reports, 2025
Wee, Kathleen, Yang, Kevin C, Schaeffer, David F, Zhou, Chen, Leung, Emily, Feng, Xiaolan, Laskin, Janessa, Marra, Marco A, Loree, Jonathan M, Gorski, Sharon M
Neuroendocrine neoplasms (NENs) encompass a highly heterogeneous group of neoplasms with varying prognoses and molecular alterations. Molecular profiling studies have furthered our understanding of NENs, but the majority of previous studies have focused on primary tumors and on mutational landscapes using DNA sequencing data. Here, we describe the genomic and transcriptomic landscapes of 28 metastatic NENs across different primary anatomical sites (PASs) and their potential clinical implications. Although our cohort is small, our analyses provide further insights on the molecular commonalities and distinctions between metastatic NENs of different PASs. Comparison to several reference transcriptome data sets revealed that despite considerable whole genome and transcriptome variability in NENs, the metastatic NENs are still more like each other than other cancer types. Our study also highlights the potential utility of NEN transcriptome data for molecular classification and clinical decision making.
Bio-protocol, 2025
Gidda, Arlene K, Chittaranjan, Suganthi, Gorski, Sharon M
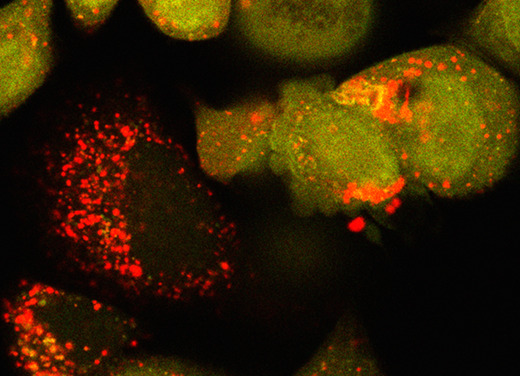
Cell viability and cytotoxicity assays are commonly used to investigate protein function and to evaluate drug efficacy in cancer and other disease models. Cytotoxicity is the measure of dead or damaged cells and is often quantified using assays based on cellular characteristics such as membrane integrity or mitochondrial metabolism. However, these assays are typically limited to endpoint analysis and lack emulation of physiological conditions. The IncuCyte Live and Dead Cell assay described here leverages common cell permeability methodologies but uses fluorescence microscopy channels to image both live and dead cells over time and phase microscopy channels to measure confluency. Cytotox green reagent is a cell membrane-impermeable dye that can only be taken up by cells with poor cell membrane integrity. NucLight rapid red dye is a cell membrane-permeable nuclear dye that can be taken up by all cells. Based on dye uptake and fluorescence intensity, the IncuCyte software can be used to analyze images for live and dead cell detection and quantification. Phase microscopy is used to determine confluency and can be further quantified using the IncuCyte software. We provide an application of this assay, using it to calculate IC{{sub}}50{{/sub}} and EC{{sub}}50{{/sub}} values for the assessment of drug efficacy. Key features • Quantify live and dead cells over time. • Determine drug IC{{sub}}50{{/sub}} and/or EC{{sub}}50{{/sub}} in 2D cell cultures. • This protocol requires the instrument IncuCyte{{sup}}®{{/sup}} S3 (or SX5) Live-Cell Analysis system and corresponding software.
PLoS biology, 2025
Samarasekera, Gayathri, Go, Nancy E, Choutka, Courtney, Xu, Jing, Takemon, Yuka, Chan, Jennifer, Chan, Michelle, Perera, Shivani, Aparicio, Samuel, Morin, Gregg B, Marra, Marco A, Chittaranjan, Suganthi, Gorski, Sharon M
Cell stress adaptation plays a key role in normal development and in various diseases including cancer. Caspases are activated in response to cell stress, and growing evidence supports their function in non-apoptotic cellular processes. A role for effector caspases in promoting stress-induced cytoprotective autophagy was demonstrated in Drosophila, but has not been explored in the context of human cells. We found a functionally conserved role for effector caspase 3 (CASP3) and caspase 7 (CASP7) in promoting starvation or proteasome inhibition-induced cytoprotective autophagy in human breast cancer cells. The loss of CASP3 and CASP7 resulted in an increase in PARP1 cleavage, reduction in LC3B and ATG7 transcript levels, and a reduction in H2AX phosphorylation, consistent with a block in autophagy and DNA damage-induced stress response pathways. Surprisingly, in non-lethal cell stress conditions, CASP7 underwent non-canonical processing at two calpain cleavage sites flanking a PARP1 exosite, resulting in stable CASP7-p29/p30 fragments. Expression of CASP7-p29/p30 fragment(s) could rescue H2AX phosphorylation in the CASP3 and CASP7 double knockout background. Strikingly, yet consistent with these phenotypes, the loss of CASP3 and CASP7 exhibited synthetic lethality with BRCA1 loss. These findings support a role for human caspases in stress adaptation through PARP1 modulation and reveal new therapeutic avenues for investigation.
Autophagy, 2024
McMann, Emily, Gorski, Sharon M
The evolutionarily conserved ATG4 cysteine proteases regulate macroautophagy/autophagy through the priming and deconjugation of the Atg8-family proteins. In mammals there are four ATG4 family members (ATG4A, ATG4B, ATG4C, ATG4D) but ATG4D has been relatively understudied. Heightened interest in ATG4D has been stimulated by recent links to human disease. Notably, genetic variations in human were implicated in a heritable neurodevelopmental disorder. Genetic analyses in dogs, along with loss-of-function zebrafish and mouse models, further support a neuroprotective role for ATG4D. Here we discuss the evidence connecting ATG4D to neurological diseases and other pathologies and summarize its roles in both autophagy-dependent and autophagy-independent cellular processes.: ATG: autophagy related; BafA1: bafilomycin A1; BCL2: BCL2 apoptosis regulator; BH3: BCL2 homology region 3; CASP3: caspase 3; EV: extracellular vesicle; GABA: gamma aminobutyric acid; GABARAP: GABA type A receptor-associated protein; GABARAPL1: GABA type A receptor associated protein like 1; GABARAPL2: GABA type A receptor associated protein like 2; GFP: green fluorescent protein; LIR: LC3-interacting region; MAP1LC3: microtubule associated protein 1 light chain 3; MEF: mouse embryonic fibroblast; MYC: MYC proto-oncogene, bHLH transcription factor; PE: phosphatidylethanolamine; PS: phosphatidylserine; QKO: quadruple knockout; SDS-PAGE: sodium dodecyl sulfate-polyacrylamide gel; SQSTM1: sequestosome 1.
Journal of cell science, 2023
Sathiyaseelan, Paalini, Chittaranjan, Suganthi, Kalloger, Steve E, Chan, Jennifer, Go, Nancy E, Jardon, Mario A, Ho, Cally J, Hui, Theodore, Xu, Jing, Chow, Christine, Gao, Dongxia, Johnson, Fraser D, Lockwood, William W, Morin, Gregg B, Renouf, Daniel J, Schaeffer, David F, Gorski, Sharon M
Pancreatic ductal adenocarcinoma (PDAC) exhibits elevated levels of autophagy, which promote tumor progression and treatment resistance. ATG4B is an autophagy-related cysteine protease under consideration as a potential therapeutic target, but it is largely unexplored in PDAC. Here, we investigated the clinical and functional relevance of ATG4B expression in PDAC. Using two PDAC patient cohorts, we found that low ATG4B mRNA or protein expression is associated with worse patient survival outcomes, poorly differentiated PDAC tumors and a lack of survival benefit from adjuvant chemotherapy. In PDAC cell lines, ATG4B knockout reduced proliferation, abolished processing of LC3B (also known as MAP1LC3B), and reduced GABARAP and GABARAPL1 levels, but increased ATG4A levels. ATG4B and ATG4A double knockout lines displayed a further reduction in proliferation, characterized by delays in G1-S phase transition and mitosis. Pro-LC3B accumulated aberrantly at the centrosome with a concomitant increase in centrosomal proteins PCM1 and CEP131, which was rescued by exogenous ATG4B. The two-stage cell cycle defects following ATG4B and ATG4A loss have important therapeutic implications for PDAC.
Autophagy, 2022
Xu, Jing, Yang, Kevin C, Go, Nancy Erro, Colborne, Shane, Ho, Cally J, Hosseini-Beheshti, Elham, Lystad, Alf H, Simonsen, Anne, Guns, Emma Tomlinson, Morin, Gregg B, Gorski, Sharon M
Chloroquine (CQ), a lysosomotropic agent, is commonly used to inhibit lysosomal degradation and macroautophagy/autophagy. Here we investigated the cell-extrinsic effects of CQ on secretion. We showed that lysosomal and autophagy inhibition by CQ altered the secretome, and induced the release of Atg8 orthologs and autophagy receptors. Atg8-family proteins, in particular, were secreted inside small extracellular vesicles (sEVs) in a lipidation-dependent manner. CQ treatment enhanced the release of Atg8-family proteins inside sEVs. Using full-length ATG16L1 and an ATG16L1 mutant that enables Atg8-family protein lipidation on double but not on single membranes, we demonstrated that LC3B is released in two distinct sEV populations: one enriched with SDCBP/Syntenin-1, CD63, and endosomal lipidated LC3B, and another that contains LC3B but is not enriched with SDCBP/Syntenin-1 or CD63, and which our data supports as originating from a double-membrane source. Our findings underscore the context-dependency of sEV heterogeneity and composition, and illustrate the integration of autophagy and sEV composition in response to lysosomal inhibition. ACTB: actin beta; ANOVA: analysis of variance; ATG4B: autophagy related 4B cysteine peptidase; Atg8: autophagy related 8; ATG16L1: autophagy related 16 like 1; ATP5F1A/ATP5a: ATP synthase F1 subunit alpha; CALCOCO2: calcium binding and coiled-coil domain 2; CASP3: caspase 3; CASP7: caspase 7; CQ: chloroquine; CD9: CD9 molecule; CD63: CD63 molecule; DAPI: 4',6-diamidino-2-phenylindole; DQ-BSA: dye quenched-bovine serum albumin; ER: endoplasmic reticulum; ERN1/IRE1a: endoplasmic reticulum to nucleus signaling 1; EV: extracellular vesicles; FBS: fetal bovine serum; FDR: false discovery rate; GABARAP: GABA type A receptor-associated protein; GABARAPL2: GABA type A receptor associated protein like 2; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; GFP: green fluorescent protein; GO: gene ontology; HCQ: hydroxychloroquine; HSP90AA1: heat shock protein 90 alpha family class A member 1; IP: immunoprecipitation; KO: knockout; LAMP2: lysosomal associated membrane protein 2; LIR: LC3-interacting region; LMNA: lamin A/C; MAP1LC3B/LC3B: microtubule associated protein 1 light chain 3 beta; MS: mass spectrometry; NBR1: NBR1 autophagy cargo receptor; NCOA4: nuclear receptor coactivator 4; NTA: nanoparticle tracking analysis; PE: phosphatidylethanolamine; PECA: probe-level expression change averaging; SDCBP/syntenin-1: syndecan binding protein; SD: standard deviation; SE: secreted; sEV: small extracellular vesicles; SQSTM1/p62: sequestosome 1; TAX1BP1: Tax1 binding protein 1; TEM: transmission electron microscopy; TMT: tandem-mass tag; TSG101: tumor susceptibility 101; ULK1: unc-51 like autophagy activating kinase 1; WC: whole cell.
Biologia futura, 2022
Chan, Jennifer C Y, Gorski, Sharon M
GABARAPL2 was initially characterized for its involvement in protein transport and membrane fusion events, but has since gained notoriety for its role in autophagy. GABARAPL2 is frequently studied alongside its GABARAP subfamily members, GABARAP and GABARAPL1. Although functional redundancy exists among the subfamily members, a complex network of molecular interactions, physiological processes and pathologies can be primarily related to GABARAPL2. GABARAPL2 has a multifaceted role, ranging from cellular differentiation to intracellular degradation. Much of what we know about GABARAPL2 is gained through identifying its interacting partners-a list that is constantly growing. In this article, we review both the autophagy-dependent and autophagy-independent roles of GABARAPL2, and emphasize their implications for both health and disease.
STAR protocols, 2022
Yang, Kevin C, Gorski, Sharon M
RNA-sequencing and quantitative proteomic profiling simultaneously measure thousands of molecules and provide opportunities to decipher the transcriptomic and proteomic landscapes of cohort specimens for basic and health research. We present a protocol for the analysis of paired transcriptome and proteome data to identify and compare molecular subgroups among cohort specimens. We demonstrate a streamlined analysis workflow, applicable for both transcriptome and proteome data, which allows the comparison of two data types for RNA-protein variations and for derivation of biological implications. For complete details on the use and execution of this protocol, please refer to Yang et al. (2021).
Cell reports, 2021
Yang, Kevin C, Kalloger, Steve E, Aird, John J, Lee, Michael K C, Rushton, Christopher, Mungall, Karen L, Mungall, Andrew J, Gao, Dongxia, Chow, Christine, Xu, Jing, Karasinska, Joanna M, Colborne, Shane, Jones, Steven J M, Schrader, Jörg, Morin, Ryan D, Loree, Jonathan M, Marra, Marco A, Renouf, Daniel J, Morin, Gregg B, Schaeffer, David F, Gorski, Sharon M
Pancreatic neuroendocrine neoplasms (PNENs) are biologically and clinically heterogeneous. Here, we use a multi-omics approach to uncover the molecular factors underlying this heterogeneity. Transcriptomic analysis of 84 PNEN specimens, drawn from two cohorts, is substantiated with proteomic profiling and identifies four subgroups: Proliferative, PDX1-high, Alpha cell-like and Stromal/Mesenchymal. The Proliferative subgroup, consisting of both well- and poorly differentiated specimens, is associated with inferior overall survival probability. The PDX1-high and Alpha cell-like subgroups partially resemble previously described subtypes, and we further uncover distinctive metabolism-related features in the Alpha cell-like subgroup. The Stromal/Mesenchymal subgroup exhibits molecular characteristics of YAP1/WWTR1(TAZ) activation suggestive of Hippo signaling pathway involvement in PNENs. Whole-exome sequencing reveals subgroup-enriched mutational differences, supported by activity inference analysis, and identifies hypermorphic proto-oncogene variants in 14.3% of sequenced PNENs. Our study reveals differences in cellular signaling axes that provide potential directions for PNEN patient stratification and treatment strategies.
Autophagy, 2021
Ho, Cally J, Samarasekera, Gayathri, Rothe, Katharina, Xu, Jing, Yang, Kevin C, Leung, Emily, Chan, Michelle, Jiang, Xiaoyan, Gorski, Sharon M
Proteome profiling and global protein-interaction approaches have significantly improved our knowledge of the protein interactomes of autophagy and other cellular stress-response pathways. New discoveries regarding protein complexes, interaction partners, interaction domains, and biological roles of players that are part of these pathways are emerging. The fourth Vancouver Autophagy Symposium showcased research that expands our understanding of the protein interaction networks and molecular mechanisms underlying autophagy and other cellular stress responses in the context of distinct stressors. In the keynote presentation, Dr. Wade Harper described his team's recent discovery of a novel reticulophagy receptor for selective autophagic degradation of the endoplasmic reticulum, and discussed molecular mechanisms involved in ribophagy and non-autophagic ribosomal turnover. In other presentations, both omic and targeted approaches were used to reveal molecular players of other cellular stress responses including amyloid body and stress granule formation, anastasis, and extracellular vesicle biogenesis. Additional topics included the roles of autophagy in disease pathogenesis, autophagy regulatory mechanisms, and crosstalk between autophagy and cellular metabolism in anti-tumor immunity. The relationship between autophagy and other cell stress responses remains a relatively unexplored area in the field, with future investigations required to understand how the various processes are coordinated and connected in cells and tissues. A-bodies: amyloid bodies; ACM: amyloid-converting motif; AMFR/gp78: autocrine motility factor receptor; ATG: autophagy-related; ATG4B: autophagy related 4B cysteine peptidase; CALCOCO2/NDP52: calcium binding and coiled-coil domain 2; CAR T: chimeric antigen receptor T; CASP3: caspase 3; CCPG1: cell cycle progression 1; CAR: chimeric antigen receptor; CML: chronic myeloid leukemia; CCOCs: clear cell ovarian cancers; CVB3: coxsackievirus B3; CRISPR-Cas9: clustered regularly interspaced short palindromic repeats-CRISPR associated protein 9; DDXs: DEAD-box helicases; EIF2S1/EIF-2alpha: eukaryotic translation initiation factor 2 subunit alpha; EIF2AK3: eukaryotic translation initiation factor 2 alpha kinase 3; ER: endoplasmic reticulum; EV: extracellular vesicle; FAO: fatty acid oxidation; GABARAP: GABA type A receptor-associated protein; ILK: integrin linked kinase; ISR: integrated stress response; MTOR: mechanistic target of rapamycin kinase; MPECs: memory precursory effector T cells; MAVS: mitochondrial antiviral signaling protein; NBR1: NBR1 autophagy cargo receptor; PI4KB/PI4KIIIβ: phosphatidylinositol 4-kinase beta; PLEKHM1: pleckstrin homology and RUN domain containing M1; RB1CC1: RB1 inducible coiled-coil 1; RTN3: reticulon 3; rIGSRNAs: ribosomal intergenic noncoding RNAs; RPL29: ribosomal protein L29; RPS3: ribosomal protein S3; ; sEV: small extracellular vesicles; ; SQSTM1: sequestosome 1; SF3B1: splicing factor 3b subunit 1; SILAC-MS: stable isotope labeling with amino acids in cell culture-mass spectrometry; SNAP29: synaptosome associated protein 29; TEX264: testis expressed 264, ER-phagy receptor; TNBC: triple-negative breast cancer; ULK1: unc-51 like autophagy activating kinase 1; VAS: Vancouver Autophagy Symposium.
Scientific reports, 2021
Lebovitz, Chandra, Wretham, Nicole, Osooly, Maryam, Milne, Katy, Dash, Tia, Thornton, Shelby, Tessier-Cloutier, Basile, Sathiyaseelan, Paalini, Bortnik, Svetlana, Go, Nancy Erro, Halvorsen, Elizabeth, Cederberg, Rachel A, Chow, Norman, Dos Santos, Nancy, Bennewith, Kevin L, Nelson, Brad H, Bally, Marcel B, Lam, Wan L, Gorski, Sharon M
Pathological links between neurodegenerative disease and cancer are emerging. LRRK2 overactivity contributes to Parkinson's disease, whereas our previous analyses of public cancer patient data revealed that decreased LRRK2 expression is associated with lung adenocarcinoma (LUAD). The clinical and functional relevance of LRRK2 repression in LUAD is unknown. Here, we investigated associations between LRRK2 expression and clinicopathological variables in LUAD patient data and asked whether LRRK2 knockout promotes murine lung tumorigenesis. In patients, reduced LRRK2 was significantly associated with ongoing smoking and worse survival, as well as signatures of less differentiated LUAD, altered surfactant metabolism and immunosuppression. We identified shared transcriptional signals between LRRK2-low LUAD and postnatal alveolarization in mice, suggesting aberrant activation of a developmental program of alveolar growth and differentiation in these tumors. In a carcinogen-induced murine lung cancer model, multiplex IHC confirmed that LRRK2 was expressed in alveolar type II (AT2) cells, a main LUAD cell-of-origin, while its loss perturbed AT2 cell morphology. LRRK2 knockout in this model significantly increased tumor initiation and size, demonstrating that loss of LRRK2, a key Parkinson's gene, promotes lung tumorigenesis.
Breast cancer research and treatment, 2020
Bortnik, Svetlana, Tessier-Cloutier, Basile, Leung, Samuel, Xu, Jing, Asleh, Karama, Burugu, Samantha, Magrill, Jamie, Greening, Kendall, Derakhshan, Fatemeh, Yip, Stephen, Ng, Tony, Gelmon, Karen A, Nielsen, Torsten O, Gorski, Sharon M
Previous studies indicate that breast cancer molecular subtypes differ with respect to their dependency on autophagy, but our knowledge of the differential expression and prognostic significance of autophagy-related biomarkers in breast cancer is limited.
Cancers, 2019
Ho, Cally J, Gorski, Sharon M
Despite advances in diagnostic tools and therapeutic options, treatment resistance remains a challenge for many cancer patients. Recent studies have found evidence that autophagy, a cellular pathway that delivers cytoplasmic components to lysosomes for degradation and recycling, contributes to treatment resistance in different cancer types. A role for autophagy in resistance to chemotherapies and targeted therapies has been described based largely on associations with various signaling pathways, including MAPK and PI3K/AKT signaling. However, our current understanding of the molecular mechanisms underlying the role of autophagy in facilitating treatment resistance remains limited. Here we provide a comprehensive summary of the evidence linking autophagy to major signaling pathways in the context of treatment resistance and tumor progression, and then highlight recently emerged molecular mechanisms underlying autophagy and the p62/KEAP1/NRF2 and FOXO3A/PUMA axes in chemoresistance.
Autophagy, 2019
Sathiyaseelan, Paalini, Rothe, Katharina, Yang, Kevin C, Xu, Jing, Chow, Norman S, Bortnik, Svetlana, Choutka, Courtney, Ho, Cally, Jiang, Xiaoyan, Gorski, Sharon M
In its third edition, the Vancouver Autophagy Symposium presented a platform for vibrant discussion on the differential roles of macroautophagy/autophagy in disease. This one-day symposium was held at the BC Cancer Research Centre in Vancouver, BC, bringing together experts in cell biology, protein biochemistry and medicinal chemistry across several different disease models and model organisms. The Vancouver Autophagy Symposium featured 2 keynote speakers that are well known for their seminal contributions to autophagy research, Dr. David Rubinsztein (Cambridge Institute for Medical Research) and Dr. Kay F. Macleod (University of Chicago). Key discussions included the context-dependent roles and mechanisms of dysregulation of autophagy in diseases and the corresponding need to consider context-dependent autophagy modulation strategies. Additional highlights included the differential roles of bulk autophagy versus selective autophagy, novel autophagy regulators, and emerging chemical tools to study autophagy inhibition. Interdisciplinary discussions focused on addressing questions such as which stage of disease to target, which type of autophagy to target and which component to target for autophagy modulation. : AD: Alzheimer disease; AMFR/Gp78: autocrine motility factor receptor; CCCP: carbonyl cyanide -chlorophenylhydrazone; CML: chronic myeloid leukemia; CVB3: coxsackievirus B3; DRPLA: dentatorubral-pallidoluysian atrophy; ER: endoplasmic reticulum; ERAD: ER-associated degradation; FA: focal adhesion; HCQ: hydroxychloroquine; HD: Huntingtin disease; HIF1A/Hif1α: hypoxia inducible factor 1 subunit alpha; HTT: huntingtin; IM: imatinib mesylate; MAP1LC3B: microtubule associated protein 1 light chain 3 beta; NBR1: neighbour of BRCA1; OGA: O-GlcNAcase; PDAC: pancreatic ductal adenocarcinoma; PLEKHM1: pleckstrin homology and RUN domain containing M1; polyQ: poly-glutamine; ROS: reactive oxygen species; RP: retinitis pigmentosa; SNAP29: synaptosome associated protein 29; SPCA3: spinocerebellar ataxia type 3; TNBC: triple-negative breast cancer.
Journal of cell science, 2018
Xu, Jing, Camfield, Robert, Gorski, Sharon M
The eukaryotic endomembrane system is a complex series of interconnected membranous organelles that play important roles in responding to stress and maintaining cell homeostasis during health and disease. Two components of this system, exosome biogenesis and autophagy, are linked by the endolysosomal pathway. Exosomes are cargo-laden extracellular vesicles that arise from endosome-derived multivesicular bodies, and autophagy is a lysosomal-dependent degradation and recycling pathway. Recent studies have revealed shared molecular machinery between exosome biogenesis and autophagy, as well as substantial crosstalk between these two processes. In this Review, we first describe the classic view of exosome biogenesis and autophagy, including their links to the endolysosomal pathway. We then present the evidence for autophagy-related proteins in exosome biogenesis, the emerging roles of amphisomes and the evolving models of exosome-autophagy pathway interactions. Finally, we discuss the implications of exosome and autophagy interplay in the context of neurodegeneration and cancer.
Scientific reports, 2018
Bosc, D, Vezenkov, L, Bortnik, S, An, J, Xu, J, Choutka, C, Hannigan, A M, Kovacic, S, Loo, S, Clark, P G K, Chen, G, Guay-Ross, R N, Yang, K, Dragowska, W H, Zhang, F, Go, N E, Leung, A, Honson, N S, Pfeifer, T A, Gleave, M, Bally, M, Jones, S J, Gorski, S M, Young, R N
The cysteine protease ATG4B is a key component of the autophagy machinery, acting to proteolytically prime and recycle its substrate MAP1LC3B. The roles of ATG4B in cancer and other diseases appear to be context dependent but are still not well understood. To help further explore ATG4B functions and potential therapeutic applications, we employed a chemical biology approach to identify ATG4B inhibitors. Here, we describe the discovery of 4-28, a styrylquinoline identified by a combined computational modeling, in silico screening, high content cell-based screening and biochemical assay approach. A structure-activity relationship study led to the development of a more stable and potent compound LV-320. We demonstrated that LV-320 inhibits ATG4B enzymatic activity, blocks autophagic flux in cells, and is stable, non-toxic and active in vivo. These findings suggest that LV-320 will serve as a relevant chemical tool to study the various roles of ATG4B in cancer and other contexts.
Biochemical Society transactions, 2018
Yang, Kevin C, Sathiyaseelan, Paalini, Ho, Cally, Gorski, Sharon M
Autophagy is an evolutionarily conserved lysosome-mediated degradation and recycling process, which functions in cellular homeostasis and stress adaptation. The process is highly dynamic and involves autophagosome synthesis, cargo recognition and transport, autophagosome-lysosome fusion, and cargo degradation. The multistep nature of autophagy makes it challenging to quantify, and it is important to consider not only the number of autophagosomes within a cell but also the autophagic degradative activity. The rate at which cargos are recognized, segregated, and degraded through the autophagy pathway is defined as autophagic flux. In practice, methods to measure autophagic flux typically evaluate the lysosome-mediated cargo degradation step by leveraging known autophagy markers such as MAP1LC3B (microtubule-associated proteins 1A/1B light chain 3 beta) or lysosome-dependent fluorescent agents. In this review, we summarize the tools and methods used in mammalian cultured cells pertaining to these two approaches, and highlight innovations that have led to their evolution in recent years. We also discuss the potential limitations of these approaches and recommend using a combination of strategies and multiple different autophagy markers to reliably evaluate autophagic flux in mammalian cells.
Cold Spring Harbor molecular case studies, 2018
Wong, Hui-Li, Yang, Kevin C, Shen, Yaoqing, Zhao, Eric Y, Loree, Jonathan M, Kennecke, Hagen F, Kalloger, Steve E, Karasinska, Joanna M, Lim, Howard J, Mungall, Andrew J, Feng, Xiaolan, Davies, Janine M, Schrader, Kasmintan, Zhou, Chen, Karsan, Aly, Jones, Steven J M, Laskin, Janessa, Marra, Marco A, Schaeffer, David F, Gorski, Sharon M, Renouf, Daniel J
Pancreatic neuroendocrine tumors (PNETs) are a genomically and clinically heterogeneous group of pancreatic neoplasms often diagnosed with distant metastases. Recurrent somatic mutations, chromosomal aberrations, and gene expression signatures in PNETs have been described, but the clinical significance of these molecular changes is still poorly understood, and the clinical outcomes of PNET patients remain highly variable. To help identify the molecular factors that contribute to PNET progression and metastasis, and as part of an ongoing clinical trial at the BC Cancer Agency (clinicaltrials.gov ID: NCT02155621), the genomic and transcriptomic profiles of liver metastases from five patients (four PNETs and one neuroendocrine carcinoma) were analyzed. In four of the five cases, we identified biallelic loss of and as well as recurrent regions with loss of heterozygosity. Several novel findings were observed, including focal amplification of concomitant with loss of and in one sample with wild-type and Transcriptome analyses revealed up-regulation of target genes in this sample, confirming a -driven gene expression signature. We also identified a germline fusion event in one sample that resulted in a striking C>T mutation signature profile not previously reported in PNETs. These varying molecular alterations suggest different cellular pathways may contribute to PNET progression, consistent with the heterogeneous clinical nature of this disease. Furthermore, genomic profiles appeared to correlate well with treatment response, lending support to the role of prospective genotyping efforts to guide therapy in PNETs.
Autophagy, 2017
Choutka, Courtney, DeVorkin, Lindsay, Go, Nancy Erro, Hou, Ying-Chen Claire, Moradian, Annie, Morin, Gregg B, Gorski, Sharon M
The 2 main degradative pathways that contribute to proteostasis are the ubiquitin-proteasome system and autophagy but how they are molecularly coordinated is not well understood. Here, we demonstrate an essential role for an effector caspase in the activation of compensatory autophagy when proteasomal activity is compromised. Functional loss of Hsp83, the Drosophila ortholog of human HSP90 (heat shock protein 90), resulted in reduced proteasomal activity and elevated levels of the effector caspase Dcp-1. Surprisingly, genetic analyses showed that the caspase was not required for cell death in this context, but instead was essential for the ensuing compensatory autophagy, female fertility, and organism viability. The zymogen pro-Dcp-1 was found to interact with Hsp83 and undergo proteasomal regulation in an Hsp83-dependent manner. Our work not only reveals unappreciated roles for Hsp83 in proteasomal activity and regulation of Dcp-1, but identifies an effector caspase as a key regulatory factor for sustaining adaptation to cell stress in vivo.
Oncotarget, 2016
Bortnik, Svetlana, Choutka, Courtney, Horlings, Hugo M, Leung, Samuel, Baker, Jennifer H, Lebovitz, Chandra, Dragowska, Wieslawa H, Go, Nancy E, Bally, Marcel B, Minchinton, Andrew I, Gelmon, Karen A, Gorski, Sharon M
Autophagy, a lysosome-mediated degradation and recycling process, functions in advanced malignancies to promote cancer cell survival and contribute to cancer progression and drug resistance. While various autophagy inhibition strategies are under investigation for cancer treatment, corresponding patient selection criteria for these autophagy inhibitors need to be developed. Due to its central roles in the autophagy process, the cysteine protease ATG4B is one of the autophagy proteins being pursued as a potential therapeutic target. In this study, we investigated the expression of ATG4B in breast cancer, a heterogeneous disease comprised of several molecular subtypes. We examined a panel of breast cancer cell lines, xenograft tumors, and breast cancer patient specimens for the protein expression of ATG4B, and found a positive association between HER2 and ATG4B protein expression. We showed that HER2-positive cells, but not HER2-negative breast cancer cells, require ATG4B to survive under stress. In HER2-positive cells, cytoprotective autophagy was dependent on ATG4B under both starvation and HER2 inhibition conditions. Combined knockdown of ATG4B and HER2 by siRNA resulted in a significant decrease in cell viability, and the combination of ATG4B knockdown with trastuzumab resulted in a greater reduction in cell viability compared to trastuzumab treatment alone, in both trastuzumab-sensitive and -resistant HER2 overexpressing breast cancer cells. Together these results demonstrate a novel association of ATG4B positive expression with HER2 positive breast cancers and indicate that this subtype is suitable for emerging ATG4B inhibition strategies.
Autophagy, 2015
Lebovitz, Chandra B, Robertson, A Gordon, Goya, Rodrigo, Jones, Steven J, Morin, Ryan D, Marra, Marco A, Gorski, Sharon M
Aberrant activation or disruption of autophagy promotes tumorigenesis in various preclinical models of cancer, but whether the autophagy pathway is a target for recurrent molecular alteration in human cancer patient samples is unknown. To address this outstanding question, we surveyed 211 human autophagy-associated genes for tumor-related alterations to DNA sequence and RNA expression levels and examined their association with patient survival outcomes in multiple cancer types with sequence data from The Cancer Genome Atlas consortium. We found 3 (RB1CC1/FIP200, ULK4, WDR45/WIPI4) and one (ATG7) core autophagy genes to be under positive selection for somatic mutations in endometrial carcinoma and clear cell renal carcinoma, respectively, while 29 autophagy regulators and pathway interactors, including previously identified KEAP1, NFE2L2, and MTOR, were significantly mutated in 6 of the 11 cancer types examined. Gene expression analyses revealed that GABARAPL1 and MAP1LC3C/LC3C transcripts were less abundant in breast cancer and non-small cell lung cancers than in matched normal tissue controls; ATG4D transcripts were increased in lung squamous cell carcinoma, as were ATG16L2 transcripts in kidney cancer. Unsupervised clustering of autophagy-associated mRNA levels in tumors stratified patient overall survival in 3 of 9 cancer types (acute myeloid leukemia, clear cell renal carcinoma, and head and neck cancer). These analyses provide the first comprehensive resource of recurrently altered autophagy-associated genes in human tumors, and highlight cancer types and subtypes where perturbed autophagy may be relevant to patient overall survival.
The Journal of cell biology, 2014
DeVorkin, Lindsay, Go, Nancy Erro, Hou, Ying-Chen Claire, Moradian, Annie, Morin, Gregg B, Gorski, Sharon M
Increasing evidence reveals that a subset of proteins participates in both the autophagy and apoptosis pathways, and this intersection is important in normal physiological contexts and in pathological settings. In this paper, we show that the Drosophila effector caspase, Drosophila caspase 1 (Dcp-1), localizes within mitochondria and regulates mitochondrial morphology and autophagic flux. Loss of Dcp-1 led to mitochondrial elongation, increased levels of the mitochondrial adenine nucleotide translocase stress-sensitive B (SesB), increased adenosine triphosphate (ATP), and a reduction in autophagic flux. Moreover, we find that SesB suppresses autophagic flux during midoogenesis, identifying a novel negative regulator of autophagy. Reduced SesB activity or depletion of ATP by oligomycin A could rescue the autophagic defect in Dcp-1 loss-of-function flies, demonstrating that Dcp-1 promotes autophagy by negatively regulating SesB and ATP levels. Furthermore, we find that pro-Dcp-1 interacts with SesB in a nonproteolytic manner to regulate its stability. These data reveal a new mitochondrial-associated molecular link between nonapoptotic caspase function and autophagy regulation in vivo.