Rapid advancements in DNA sequencing have spurred the development of commercial technologies capable of enormous throughput, reducing both the amount of time and money required to sequence a genome and shifting the bottleneck from obtaining DNA sequences to assembling and analyzing them. But a new tool has now come into play, reducing both the time and computational power needed to combine the millions of pieces of a genome into one complete picture.
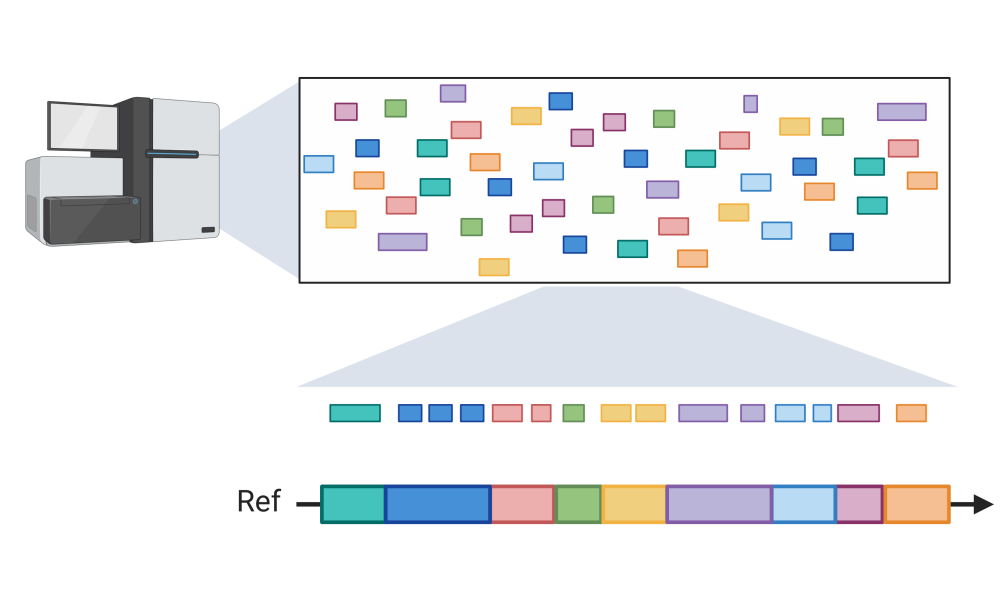
Since the advent of next generation sequencing technologies more than 15 years ago, DNA sequencers have evolved to rapidly and accurately sequence billions of bases of DNA. But they can only sequence them in short stretches, ranging from 150–250 bases using short-read sequencing technologies, and beyond a million bases using long-read sequencing instruments. For perspective, the human genome is three billion base pairs in length.
Regardless of the type of sequencing technology used, these short stretches of DNA—called reads—must be assembled to form the complete genome, analogous to a massive jigsaw puzzle that must be put together to reveal a complete picture. Long read sequencing technologies have the advantage of producing fewer pieces that must be assembled but are limited by their accuracy, which remains around 90 per cent. Short read sequencing, while much less prone to error, requires more computation to produce an assembly.
In a study published in the journal Bioinformatics led by Dr. Inanc Birol, Distinguished Scientist at Canada’s Michael Smith Genome Sciences Centre (GSC) at BC Cancer, and René Warren, Group Leader in Dr. Birol’s team, the scientists describe a new bioinformatics technology, called ntJoin, for rapid and lightweight genome assembly scaffolding.
“Having high quality genome assemblies is extremely important for many different research questions,” said Lauren Coombe, lead author on the study. “ntJoin leverages a computer science concept called minimizer sketches, which enables it to be faster and use a lot less memory than the existing state-of-the-art tools. And in most of our tests it also gives us better assembly quality with fewer mistakes.”
The tool is best suited for research applications where a genome sequence exists for a member of the same species or a closely related species. Basing an assembly on a reference genome would be similar to completing a jigsaw puzzle based on a very blurry, possibly incomplete reference picture—easier than not having a reference picture at all, but still very challenging and time consuming.
“Population sequencing studies and assemblies of genome sequences from the same individual, different individuals of the same species, and closely related species are all research goals you could use ntJoin for,” said Coombe.
ntJoin is particularly useful for hybrid assemblies—combining both short- and long-read sequencing technologies to more rapidly produce assemblies with high accuracy.
“You can use ntJoin to scaffold a highly accurate but more fragmented short read assembly using a less accurate long read assembly with longer sequences,” says Coombe. “You get the best of both worlds. You get an assembly with really long pieces and high accuracy.”
The development of ntJoin is derived from an idea and concept presented by Warren, which takes advantage of minimizer sketches and makes use of code developed for other projects in the group. Borrowing existing code from these projects enabled Coombe to get ntJoin running quickly. But designing a bioinformatics tool is like doing an experiment: just because you get an answer, it doesn’t mean it's the right one. ntJoin went through a series of iterations, each with the code slightly tweaked to produce a better result, and was rigorously tested by the group.
“These projects tend to be continuously evolving. You start with something simple and continue to implement more features to improve accuracy and utility of the tool,” said Coombe.
ntJoin is a free, open-access tool available for scientists to download and use at github.com/bcgsc/ntjoin.
Visit the GSC’s software centre.
Learn more about research at the GSC.
See other GSC publications.
Learn more about Dr. Inanc Birol’s group.
