Several neurological diseases are characterized by abnormal accumulation of short tandem repeat (STR) sequences. Today, such repeats can be detected using short- and long-read sequencing alignments. In a study published by the lab of Dr. Inanç Birol, Distinguished Scientist at the GSC, researchers have developed Straglr; a novel, robust software capable of detecting disease-causing repeats using long-read sequencing data.
Short Tandem Repeats (STRs)
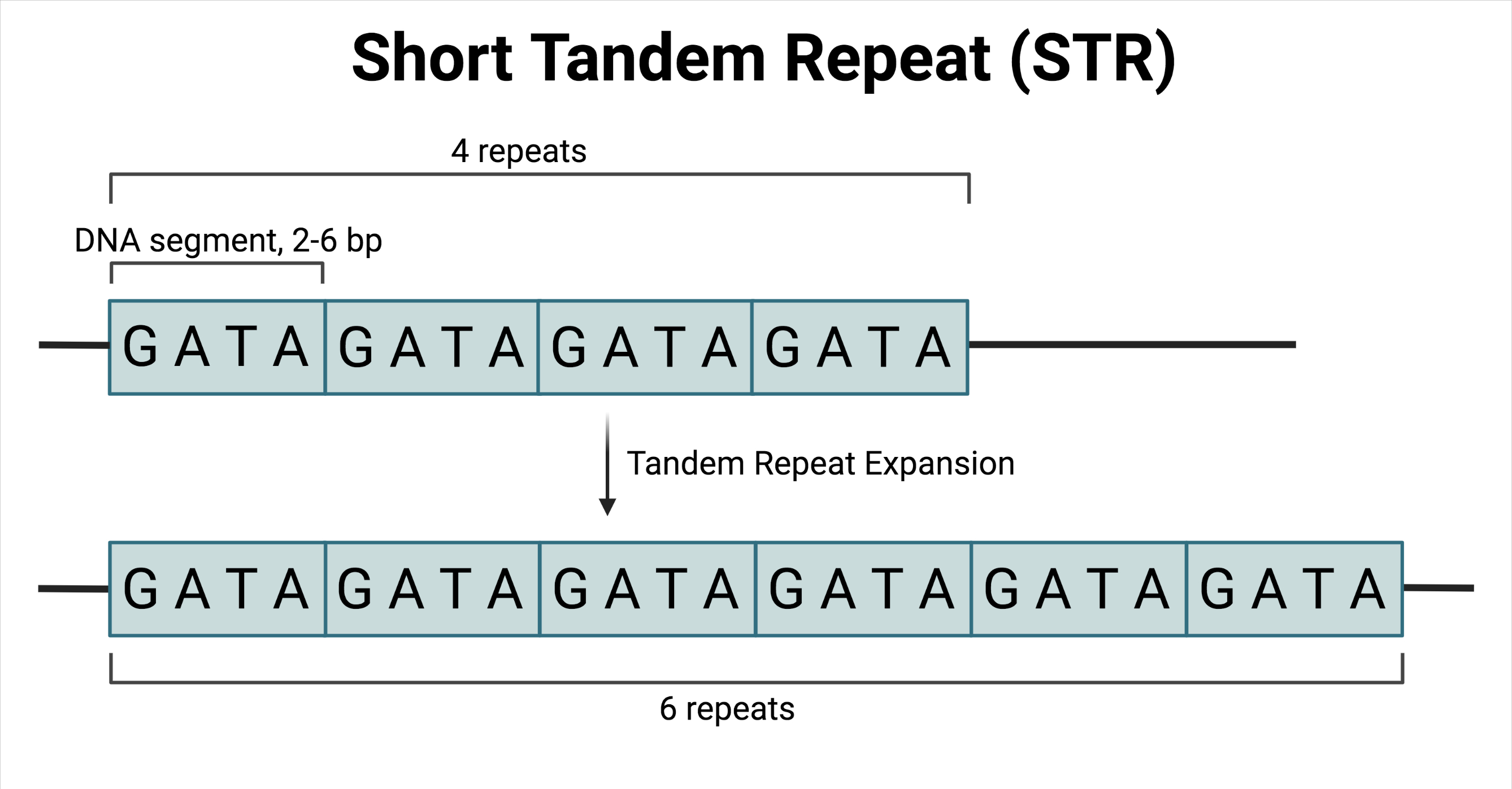
Short tandem repeats (STRs) (also referred to as “microsatellites”) are short DNA segments (individually ranging in size from two to six base pairs) that form a repeating pattern, in which multiple segments are adjacent to one another.
While typically a normal feature in a healthy human genome, an overabundance or overaccumulation of STRs has been implicated in multiple neurological disorders, including Huntington’s disease and fragile X syndrome. Such diseases are characterized by the accumulation of repeating sequences over multiple generations; a phenomenon known as tandem repeat expansion.
Incidentally, the number of repeats can be directly correlated with disease onset and heterogeneity in disease presentation, with more repeats generally indicating earlier onset and more severe disease.
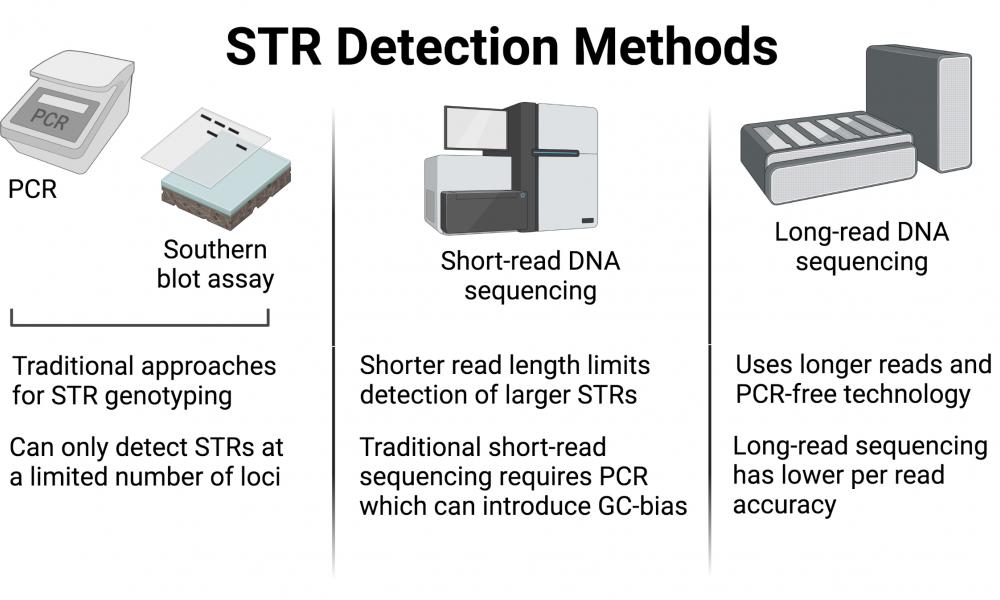
Detecting STRs
All humans have STRs of variable length at specific known locations in the genome (known as loci). While this variability has previously been exploited to differentiate or genotype individuals using traditional methods such as PCR or Southern blotting, sequencing techniques promise to be more accurate and comprehensive in characterizing STRs.
“Conventional STR detection methods look for [STRs] under the proverbial lamp-posts – known loci for which they have probes. If a clinically relevant expansion occurs beyond those loci, however, these methods are totally in the dark,” says Principal Investigator, Dr. Inanç Birol.
Today, though there are multiple ways to detect STRs, each method faces its fair share of limitations (e.g., GC-bias) and require prior knowledge about their loci. Many recent studies have explored long-read sequencing technology as a better option for detecting STRs while side-stepping many of the limitations associated with previous approaches.
“Until recently, because the read lengths were short, sequencing technologies were not a good replacement for the conventional STR detection methods,” says Dr. Birol.
Straglr, a robust software tool
Published in the journal Genome Biology, researchers in the Birol Lab have developed a new software tool, Straglr, that can be used to detect tandem repeat expansions from sequencing data acquired using PCR-free, long-read sequencing technology.
In the present study, researchers tested Straglr against other similar software by analyzing modified human reference genome sequences. To benchmark their software’s capabilities, the researchers modified the human reference genome to simulate repeat expansion at 17 loci known to be target sites for repeat expansion diseases.
Unlike other software tools, Straglr was shown to save time and computing resources, and also allowed for the discovery of repeat expansions at previously unannotated loci. With their study, the Birol Lab demonstrate the diagnostic potential for Straglr to be implemented as a tool to detect tandem repeat expansion events and also as a resource to further study the tandem repeat expansion phenomenon.
“Availability of long reads, powered with an analytical tool like Straglr, will eliminate the limitations of earlier methods and will be instrumental in broadening our understanding of the links between STR expansions and various human diseases,” says Dr. Birol.
Acknowledgements:
This study was supported by funding from the Canadian Institutes of Health Research (CIHR) and Genome Canada and Genome British Columbia.
Images created with BioRender.com.
Learn more:
Learn about other bioinformatics tools and software for analyzing sequencing data
Learn more about the Birol Lab at the GSC and about their bioinformatics technology research
Citation:
Readman Chiu, Indhu-Shree Rajan-Babu, Jan M Friedman, Inanç Birol. Straglr: Discovering and Genotyping Tandem Repeat Expansions Using Whole Genome Long-Read Sequences. Genome Biology.
*bold font indicates members of the GSC.
