When sequencing samples containing DNA from multiple species, such as when analyzing the human microbiome or identifying pathogenic organisms in an infected host, scientists use bioinformatics tools to classify the organisms present. But aligning every genome to reference can be computationally demanding and even excessive, depending on the desired data.
In these situations, “classification tools” allow computational biologists to quickly identify to which species the sequenced DNA belongs; Dr. Inanc Birol’s Bioinformatics Technology Lab (BTL) at the GSC has developed miBF, a fast classification tool with high sensitivity and specificity, and low demand on computational memory.
While classification can be done following alignment of the sequencing data to reference genomes, some research applications do not benefit from the additional information gleaned from an alignment. The need for a robust classification tool spurred the BTL to develop a more computationally efficient, alignment-free method.
A major challenge in the development of alignment-free classification tools is balancing specificity (how well the tool can handle sequencing errors) with computational costs. The most common computationally efficient tools used currently tend to suffer from low specificity. These tools work by breaking the reference and query sequences into sub-sequences of a particular length of base pairs. The low specificity of this approach can be overcome by “spaced seeds”— patterns of relevant and irrelevant positions within a sequence of certain length to improve specificity and one of the central innovations in miBF.
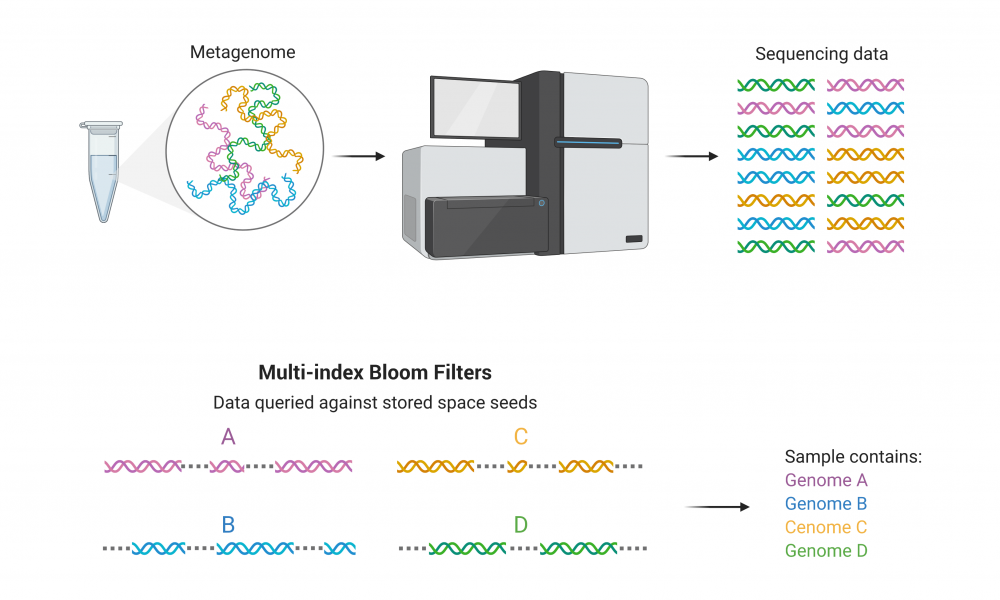
Published recently in the Proceedings of the National Academy of Science (PNAS), miBF builds upon an existing data structure known as a Bloom Filter (BF). In the PNAS study, scientists describe a novel probabilistic data structure, called a multi-index Bloom Filter, which can store multiple spaced seed sequences with a low memory cost that remains static regardless of seed length or seed design.
Incorporating and storing multiple spaced seeds sequences into the original BF data structure will enable researchers to rapidly classify samples containing DNA from not just one, but thousands of organisms with high specificity and low memory cost.
“One of the shortcomings of the basic BF data structure is that you can really ask yes or no questions against just one organism,” says Dr. Justin Chu, post-doctoral fellow in Dr. Birol’s lab and the lead author of the study. “Synergizing with spaced seeds, this new data structure allows you to query not just one, but potentially thousands of organisms.”
When compared against state-of-the-art classification tools, the researchers found that the miBF-based classifier performed with higher sensitivity and specificity while using half the memory and an order of magnitude less computational time.
miBF is freely available within BioBloom Tools through GitHub.
Learn more about the Bioinformatics Technology Lab
Learn more about software development at the GSC
