Cancers are diseases of the genome. They can result from minor changes to single or a few base pairs, known as mutations, or from larger structural changes to the genome, including how it is packaged into chromosomes and manipulated by other molecules. Understanding how things have gone wrong in specific cancers can help scientists develop better prognostic tools and treatment strategies.
DNA sequencing technology has helped scientists glean this information, but it has limitations. In a new GSC study, scientists in Dr. Steve Jones’ lab have come up with a new method to get around one such limitation.
Two copies of every gene
Every person has two copies of every gene, one from each parent, written in DNA. For many genes, both of these copies (known as alleles) are active and contribute to the growth or maintenance of a cell and organism. But for some genes only one allele is active while the other is silenced.
DNA methylation (in which a methyl group is added to segments of DNA) is one means that a cell has to silence an allele. Studying gene copy methylation patterns (known as allelic methylation) is important because any abnormality in the way that a gene is methylated can lead to disease, including cancers. But it has been very challenging, if not impossible, to study allelic methylation genome wide. That is, until now.
Long reads make a big difference

“We have developed a novel method and workflow that allows the study of allelic methylation at each gene copy using the nanopore long read sequencing technology” says Mr. Vahid Akbari, a PhD student in the Jones Lab and who is lead author on the study publication.
In this study, published in Genome Biology, Akbari, Jones and colleagues introduce new software and processes that can dramatically enhance scientists’ ability to study allelic methylation across the entire genome. Using nanopore long-read sequencing technology their study illustrates how to generate data for nearly complete allelic methylation using only a single sample, not only allowing for ease of studying multiple genes at once but also dramatically reducing the cost of sequencing and analysis. Previous studies required thousands of samples and cumbersome analytical processes to determine from which parent some silenced alleles came.
A relatively new player in the Massively Parallel Sequencing technology lineup, as its name suggest long-read sequencing allows for the sequencing of lengthy stretches of a single large molecule of DNA—up to hundreds of thousands of base pairs. In contrast, short-read technology requires DNA from many samples to be broken into thousands of fragments (known as “reads”) of a few hundred to thousand base pairs, each sequenced and then pieced together computationally. In the process, data about genes that are close together can be lost, including information about allelic methylation.
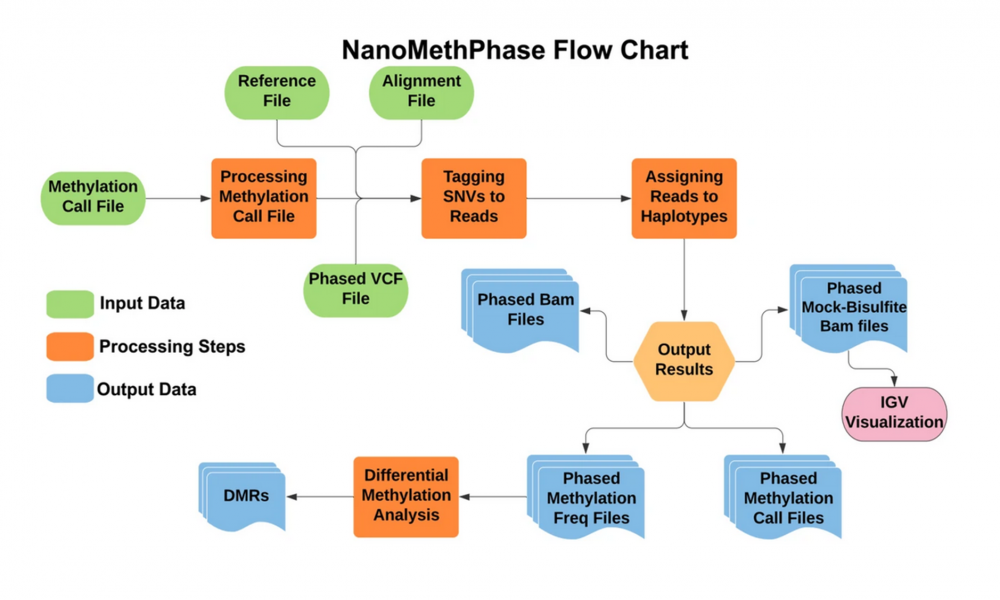
SNVoter and NanoMethPhase
“I was really surprised that nanopore allows the reliable methylation and allelic methylation detection at very low coverage—of only 10x,” says Akbari, referring to the practice of sequencing a region of the genome multiple times over to ensure data accuracy. “To get reliable methylation data from other similar technologies may require a coverage of up to 250x per strand.”
Despite the benefits, there are disadvantages to nanopore technology, mainly its high error rate for detecting small differences between genes, such as single nucleotide variants (SNV), which are also used to put the still broken long-reads together computationally. To get quality results with coverage as low as 10X challenge, the team developed new software: SNVoter and NanoMethPhase.
“SNVoter is the first post processing software for nanopore data which can considerably improve SNV calling for low coverage data and allows more accurate detection of haplotypes. And NanoMethPhase is the first tool that has been designed to leverage SNVs and methylation data to phase and match CpG methylation to their haplotypes,” says Akbari.
As for the ultimate benefit of this new software and methodology: “I am using the software and the results that come from this research to study allelic methylation in human tumor samples,” he says. “And I hope this will help us to better understand the contribution of DNA methylation in human cancer development.”
This study is supported by Canada Research Chairs and the University of British Columbia Four Year Doctoral Fellowship award.
References
Vahid Akbari, Jean-Michel Garant, Kieran O’Neill, Pawan Pandoh, Richard Moore, Marco A. Marra, Martin Hirst, Steven J. M. Jones. 2021. Megabase-scale methylation phasing using nanopore long reads and NanoMethPhase. Genome Biology 22, 68 (2021). https://doi.org/10.1186/s13059-021-02283-5
Learn more about the GSC’s technology platform.
Learn more about Sequencing at the GSC.
Learn more about Bioinformatics at the GSC.
