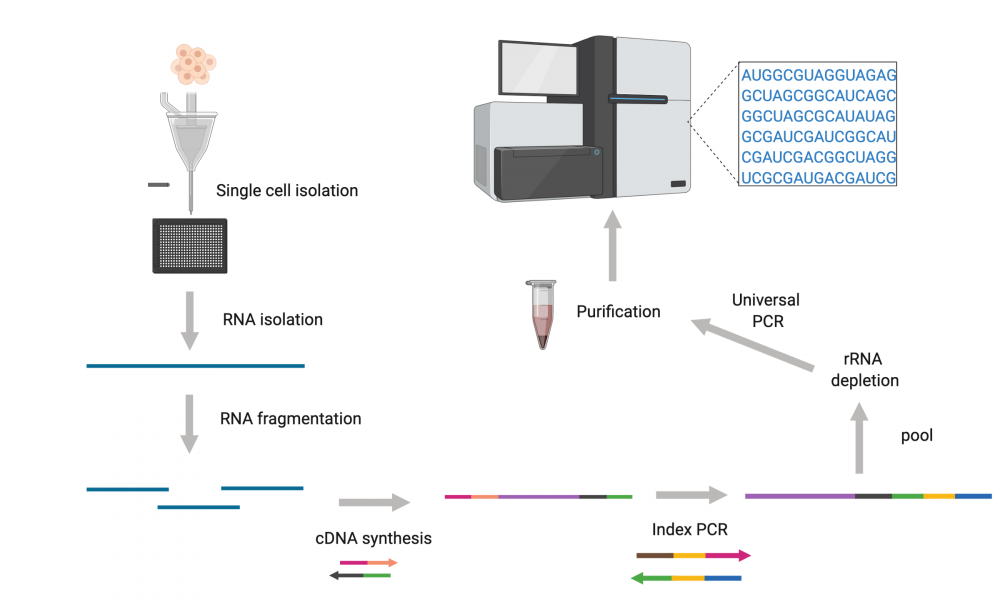
The GSC’s Technology Development team has come up with a new protocol that can sequence full-length RNA in a way that captures all classes of RNA transcripts produced by individual cells. Importantly, the adapted protocol enables sequencing of RNA transcripts from thousands of cells at a time.
Single-cell RNA sequencing (scRNAseq) is a powerful tool in cancer research because it allows scientists to study heterogeneous tissue containing multiple cell types such as tumors. Until now, however, it’s been difficult to capture the entire transcriptome of individual cells, especially at high capacity.
Limitations of scRNAseq
Prior to this study, published in Frontiers in Genetics, most scRNAseq protocols were limited to sequencing the 3’ or 5’ end of polyadenlated transcripts, allowing for quantification of gene expression but not full sequencing.
Protocols that do allow for full-length sequencing of RNA transcripts in single cells are generally limited to analysis of 96 cells at a time and like quantification methods, only capture polyadenylated transcripts (largely composed of the type of RNA that will be translated into protein).
New Protocol for scRNAseq
According to lead author, Dr. Simon Haile, the key to developing the new protocol was the ability to capture transcripts using random priming while still being able to remove rRNA-derived sequences.
“Using CRISPR-like technology, the platform we adapted uses guide RNAs at the cDNA level allowing for ribosomal RNA depletion,” says Dr. Haile.
In addition to strand-specific sequencing, the adapted protocol captures both polyadenylated and non-polyadenylated transcripts. This provides data from the complete transcriptome, including alternatively spliced and fusion transcripts. This means it captures information on non-coding RNAs that help control gene expression, most of which are not polyadenylated.
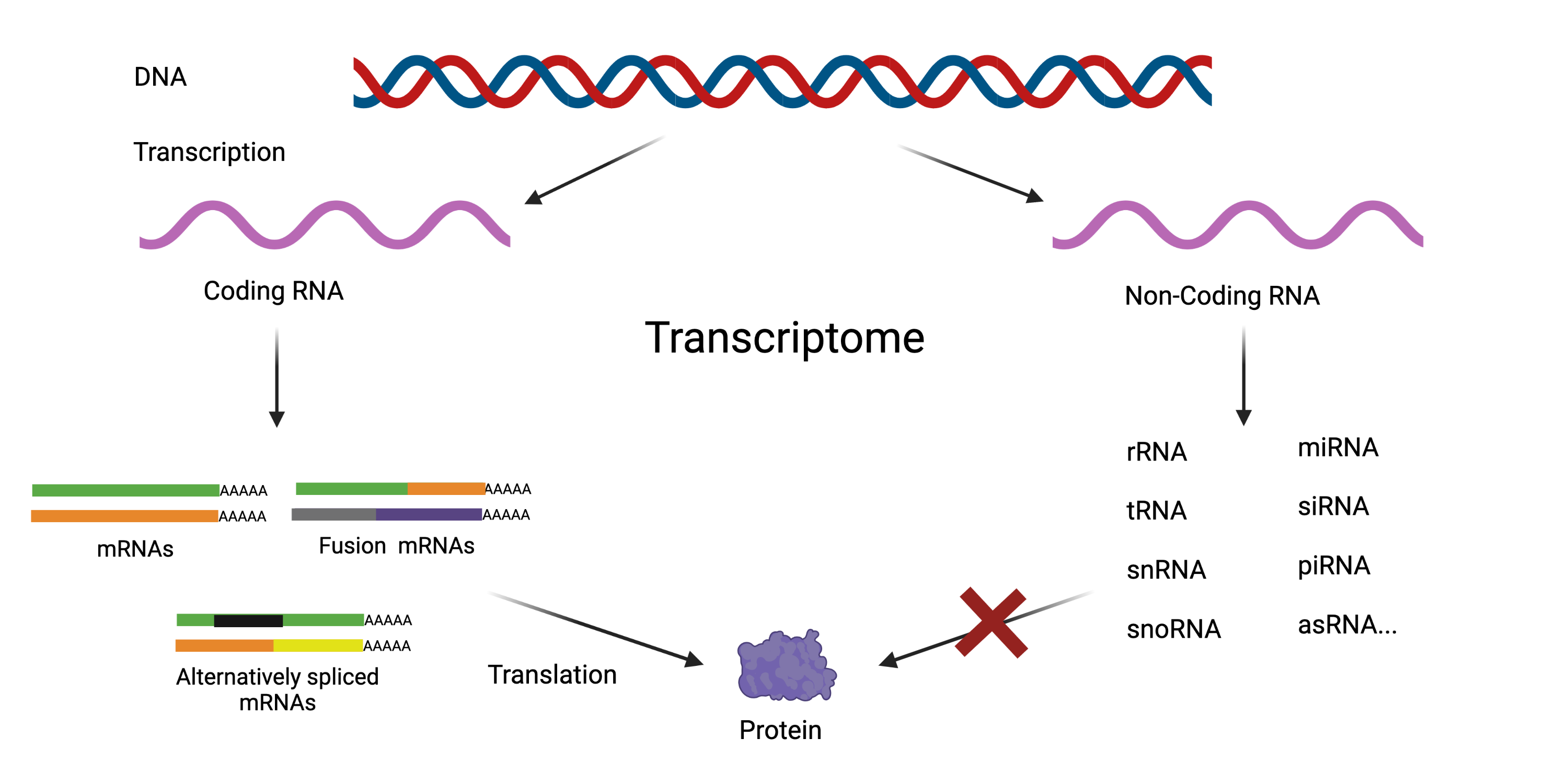 Applications of Complete scRNAseq
Applications of Complete scRNAseq
As Dr. Haile describes it, this technique allows for the examination of “how the heterogeneity of tumors change during the course of cancer treatment and disease progression. It could, for example, lead to an understanding of which cell types become prevalent as a source of treatment resistance in POG studies.”
Application of the protocol is not limited to cancer, however. The ability to fully sequence the entire transcriptome of individual cells could be used to study embryogenesis, medical microbiology or any disease where there is cell-specific development.
To further improve the methodology, members of the Technology Development Group are looking at ways to reduce the turn-around-time for results by “improving workflow to simplify the process and find ways of continuing to increase throughput.” As such, the protocol would become more cost-efficient and capable of studying a wider variety of tissues—including solid tumors—to inform clinical practice for cancer patients and beyond.
Learn more
Find out more about technology development at the GSC here.
Learn more about gene expression here.
For more detail on coding and non-coding DNA, read this review.
Citation
Simon Haile, Richard D. Corbett, Veronique G. LeBlanc, Lisa Wei, Stephen Pleasance, Steve Bilobram, Ka Ming Nip, Kirstin Brown, Eva Trinh, Jillian Smith, Diane L. Trinh, Miruna Bala, Eric Chuah, Robin J. N. Coope, Richard A. Moore, Andrew J. Mungall, Karen L. Mungall, Yongjun Zhao, Martin Hirst, Samuel Aparicio, Inanc Birol, Steven J. M. Jones, and Marco A. Marra. 03 June 2021. A Scalable Strand-Specific Protocol Enabling Full-Length Total RNA Sequencing From Single Cells. Front. Genet.
Funding
This study was funded by the Canadian Cancer Society,the Canadian Institutes of Health Research and Genome Canada/Genome British Columbia. The authors gratefully acknowledge the support of the BC Cancer Foundation, the Canada Foundation for Innovation and the Canada Research Chairs program.
Images created with BioRender.com
