
Having a high-quality assembled genome—free of errors and gaps—is important for countless biological studies. In comparison to short reads, long reads are better for sequencing highly repetitive DNA regions; however, long reads are also known for their relatively higher error rates. The Birol Lab addresses this shortcoming by developing ntEdit+Sealer, an alignment-free genome finishing protocol ideal for correcting genome assemblies generated from long read sequencing data to produce error-free, highly accurate assemblies.
Options to sequence and assemble genomes
Fully sequenced genomes are a critical tool needed for several research areas and applications including clinical settings, comparative genomics, and population studies. To fully sequence a genome, several sequencing technologies are currently available including next generation or short-read sequencing (e.g., Illumina)—also referred to as second generation sequencing—and, more recently, long-read or third generation sequencing technologies (e.g., Oxford Nanopore Technologies Ltd. and Pacific Biosciences (PacBio) of California, Inc.).
In long-read sequencing of genomes, several copies of the same genome are randomly fragmented into multiple large chunks, sequenced individually, and then pieced together by their overlapping regions and “assembled” to re-form the completed genome.
Long-read sequencing technologies are advantageous over short-reads as they are better for resolving repetitive DNA regions. However, long reads have a major limitation as they are known for having an appreciable error rate in terms of sequencing accuracy. To overcome this limitation, draft genome assemblies usually have to undergo a so-called “genome polishing step” to improve their accuracy.
Long-read genome polishing step
Some genome polishing tools (e.g., Pilon and Racon) use a “hybrid” approach where short reads (which are more accurate) are aligned to the long reads and used to identify, validate, and correct base errors in long-read genome assemblies. The approach of aligning short reads to the long-read draft genome assembly provides a lot of information about assembly quality, but can also be memory intensive and computationally costly for organisms with large genomes. To address this issue, scalable automated tools for polishing and completing long-read genome assemblies are needed.
Published recently in Current Protocols, the Birol Lab has developed ntEdit+Sealer, a scalable, alignment-free, k-mer based genome finishing protocol that utilizes short reads and memory-efficient Bloom filters to conduct genome polishing and gap filling.
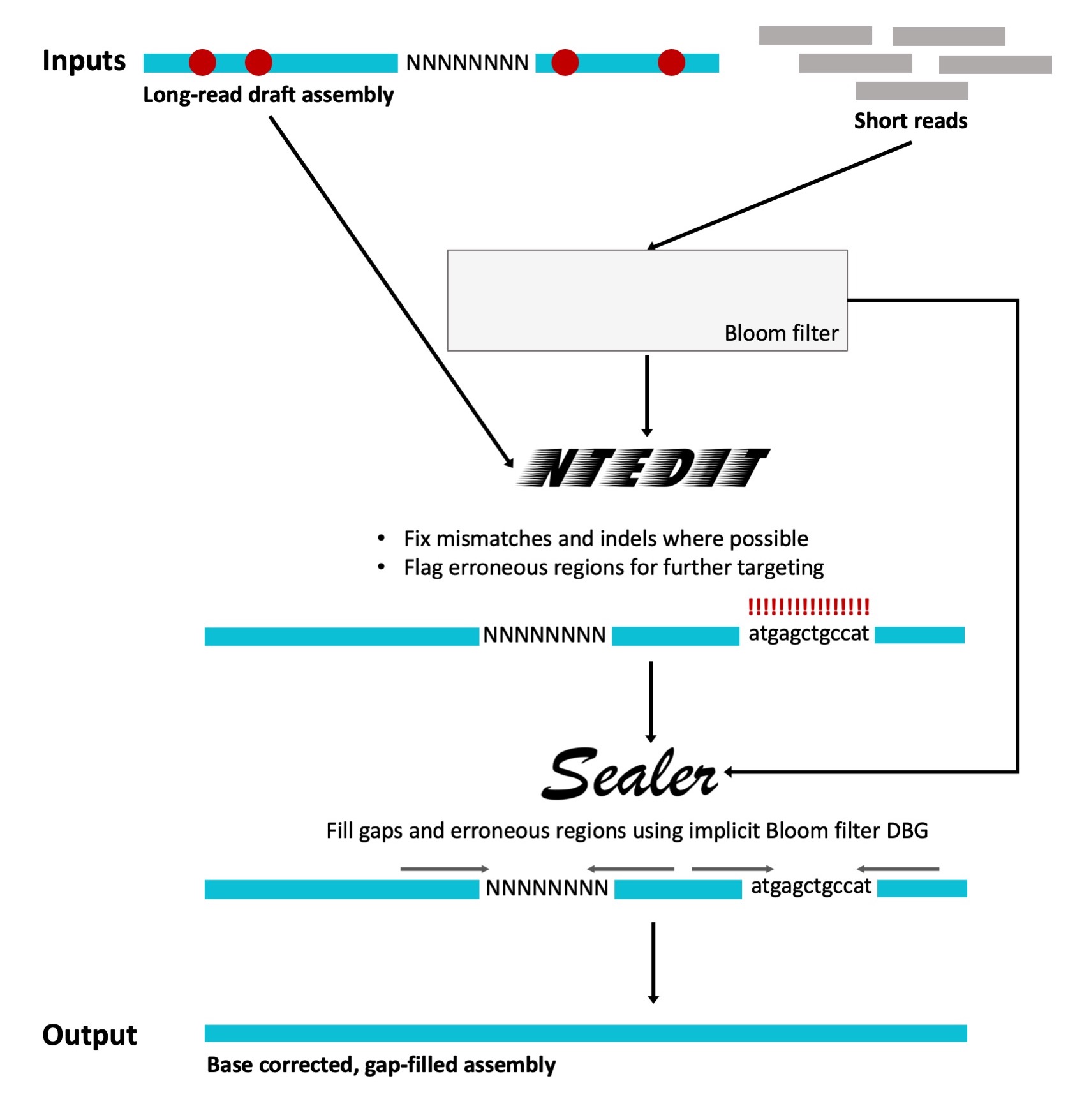
ntEdit+Sealer is composed of a duo of separately developed tools published by the Birol Lab: ntEdit1, which is used to correct local misassemblies (base mismatch and indels) and flag extended regions that require further correction, and Sealer2, which fills-in assembly gaps and problematic regions flagged by ntEdit. By combining the two tools in a multi-step approach, error correction can be done at two different levels of resolution: ntEdit resolves assembly errors on a small scale while Sealer works on the large scale (i.e., filling in the longer erroneous assembly regions and gaps). As a whole, ntEdit+Sealer produces highly accurate, error-corrected genome assemblies. Both tools—ntEdit and Sealer—use Bloom filters.
What are Bloom filters?
Bloom filters are a space-efficient probabilistic data structure designed to store elements and test whether an element is present in a set. These operations can be carried out rapidly and efficiently through the use of hash functions. For example, a Bloom filter may be used to quickly check to see if a username is available, where the set is the list of all registered usernames. Since Bloom filters are made up of an array of bits, they require less memory during runtime and storage operations.
By employing Bloom filters here, the researchers ensured that each of their tools (ntEdit and Sealer) were more memory efficient and worked faster than typical alignment-based approaches.
Discussing the innovation behind this genome polishing protocol, lead author Janet Li says: “The ntEdit+Sealer protocol uses Bloom filters, a highly efficient data structure, to identify and correct genome assembly errors at base level, as well as larger, more difficult to assemble regions. This tool is more effective and efficient than alignment-based hybrid polishers and is more accessible for groups that may have limited computational resources or time. ntEdit+Sealer is wrapped up in a pipeline that requires just a single command to run, and the published protocol provides step-by-step guidance for users with limited bioinformatics experience or know-how.”
The researchers anticipate that ntEdit+Sealer will be invaluable as a genome assembly finishing protocol for long-read draft genome assemblies with short (and soon long) sequencing reads and will be essential for producing accurate genome assemblies. ntEdit+Sealer is available as a Makefile pipeline here.
Acknowledgements:
This study was supported financially by Genome Canada and Genome British Columbia, and the National Institutes of Health.
Learn more:
1. ntEdit was published by Warren at al. (2019); learn more here
2. Sealer was published by Paulino et al. (2015); learn more here
3. Learn about other bioinformatics tools and software for analyzing sequencing data
4. Learn more about the Birol Lab at the GSC and about their bioinformatics technology research
Citation:
Li JX, Coombe L, Wong J, Birol I, Warren RL. ntEdit+Sealer: Efficient Targeted Error Resolution and Automated Finishing of Long-Read Genome Assemblies. Curr Protoc. 2022 May;2(5):e442. doi: 10.1002/cpz1.442. PMID: 35567771.
*bold font indicates members of the GSC.
