
Current sequencing methods generate information (or “reads”) that cover small pieces of non-continuous DNA. To gather information about whole genomes, overlapping reads need to be identified and merged (or “assembled”) in order to create a genome assembly. Assemblies generated from whole-genome shotgun sequencing (WGS) include many gaps, which can affect downstream analyses. GSC scientists have developed a new tool, called Physlr, that makes use of linked-read sequencing data to create next-generation physical maps with fewer gaps that will facilitate a wide range of applications, such as scaffolding of draft assemblies.
Sequencing methods and genome physical maps
DNA sequencing can be used to determine the nucleotide sequence of an individual’s genome; however, the genome first needs to be physically fragmented to generate DNA fragments (or molecules) of an appropriate size for sequencing. These fragments then need to be strung together in order to assemble them into a full (or nearly full) genome.
In early sequencing approaches (e.g. Sanger sequencing), physical molecule mapping methods were employed that, while highly effective, were labour-intensive, costly, and time-consuming. Whole-genome shotgun sequencing (WGS) paired with high-throughput sequencing platforms gained popularity in the mid-to-late 2000s, in part due to its lower cost.
WGS relies on random fragmentation of the genome, followed by sequencing and then computational assembly of these fragments based on their sequence overlap. Due to the short-read nature of most of these methods, the resulting assemblies are typically highly fragmented, since short reads originating from repetitive or complex regions often cannot be confidently assigned to a single area of the genome.
More recently, new sequencing technologies have emerged that provide longer-range genomic information, such as long-read and linked-read sequencing. By providing additional location context, data derived from these sequencing methods can be used to generate assemblies with fewer gaps (i.e. that are more contiguous). However, up-to-date bioinformatic tools are required to compile these data effectively.
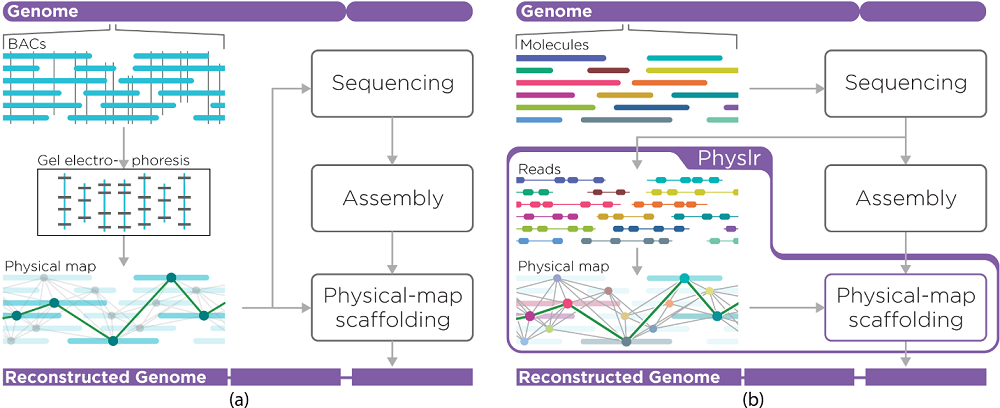
De novo assembly of linked-read data
To improve the assembly potential of linked-read sequencing data, researchers from Dr Inanç Birol’s lab at the GSC have developed a new bioinformatic tool called Physlr. Physlr leverages information from linked-read data to generate next-generation physical maps, and incorporates several novel approaches to do so.
For example, linked-read sequencing makes use of barcodes that identify molecules derived from nearby regions of DNA; however, these barcodes can be reused, meaning that they are not necessarily unique to one region. Physlr is the only tool of its kind that can separate molecules from different regions of the genome with the same barcode in a manner that is scalable to large datasets.
To achieve this, the authors devised a novel computational method for so-called “community detection”, which identifies groups of data points that are more similar to each other than they are to other groups. Community detection algorithms are used commonly across fields for various bioinformatic applications; therefore, this novel tool may also find applications in other areas of research.
High-quality assemblies enable downstream applications
Following sequencing, assembly of the resulting reads into a genomic structure is the first step in analysing sequencing data; therefore, the quality of this assembly has a major impact on the nature and quality of downstream analyses that can be performed. In their study, Afshinfard et al. demonstrated that the next-generation physical maps generated by Physlr provide high-quality scaffolds that improve the contiguity of human genome assemblies compared to other available tools.

The authors of the study point out that these improved maps will also facilitate other downstream analyses, such as the detection of structural rearrangements of the genome. Physlr can also provide information on whether mutations or other changes are on the same molecule of DNA, something that’s not always possible with less contiguous genome assemblies.
Learn more:
Learn more about ongoing research in the Bioinformatics Technology Lab, led by Dr Birol, at the GSC.
Acknowledgements:
This work was supported by Genome BC, Genome Canada, and the National Institutes of Health.
Figure 1 was created by Amirhossein Afshinfard and has been adapted for the cover of DNA (2022, 2(2)). Figure 2 is from Afshinfard et al. (DNA 2022).
Citation:
Afshinfard A, Jackman SD, Wong J, Coombe L, Chu J, Nikolic V, Dilek G, Malkoç Y, Warren RL, Birol I. Physlr: Next-Generation Physical Maps. DNA.
