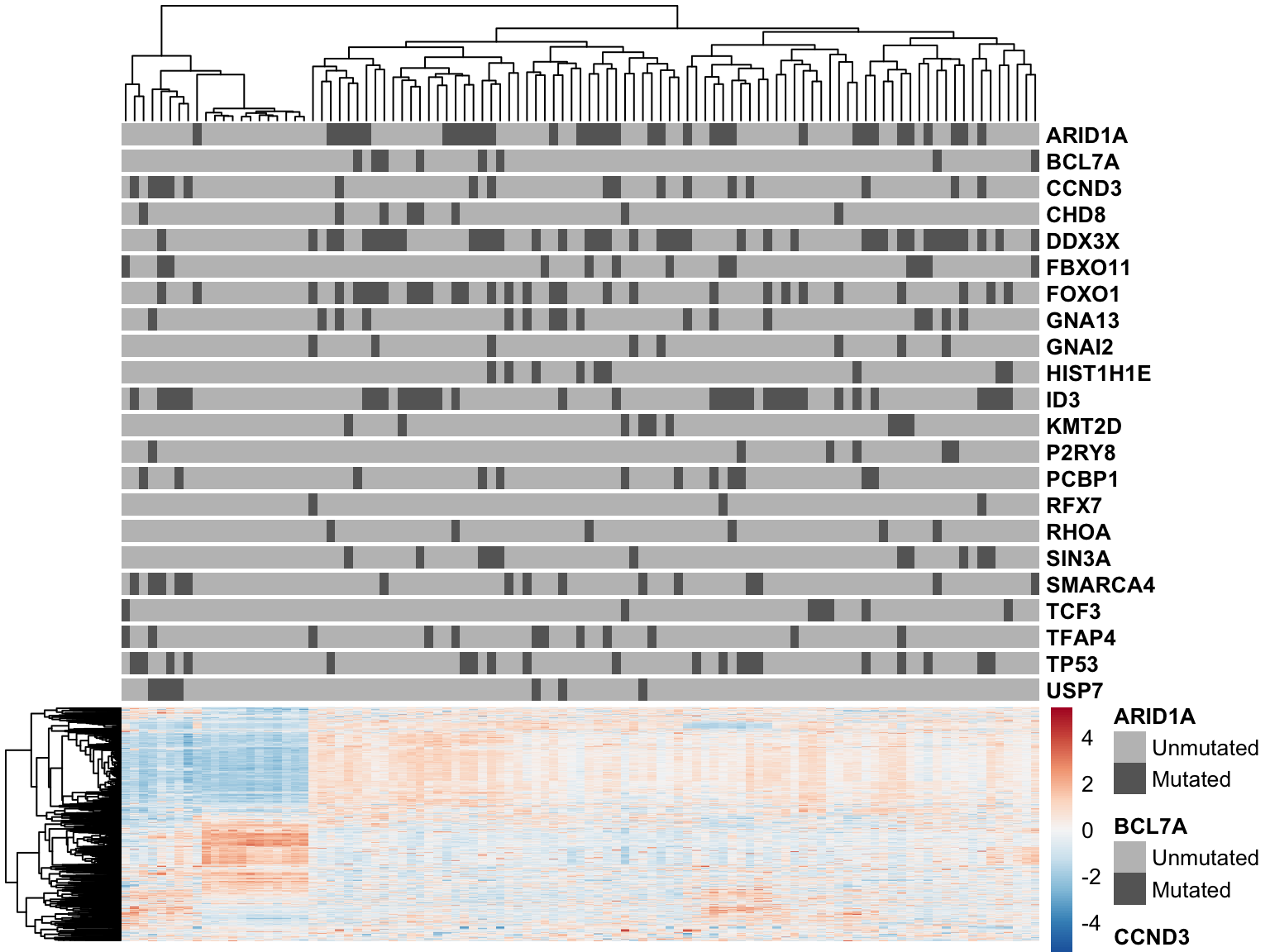
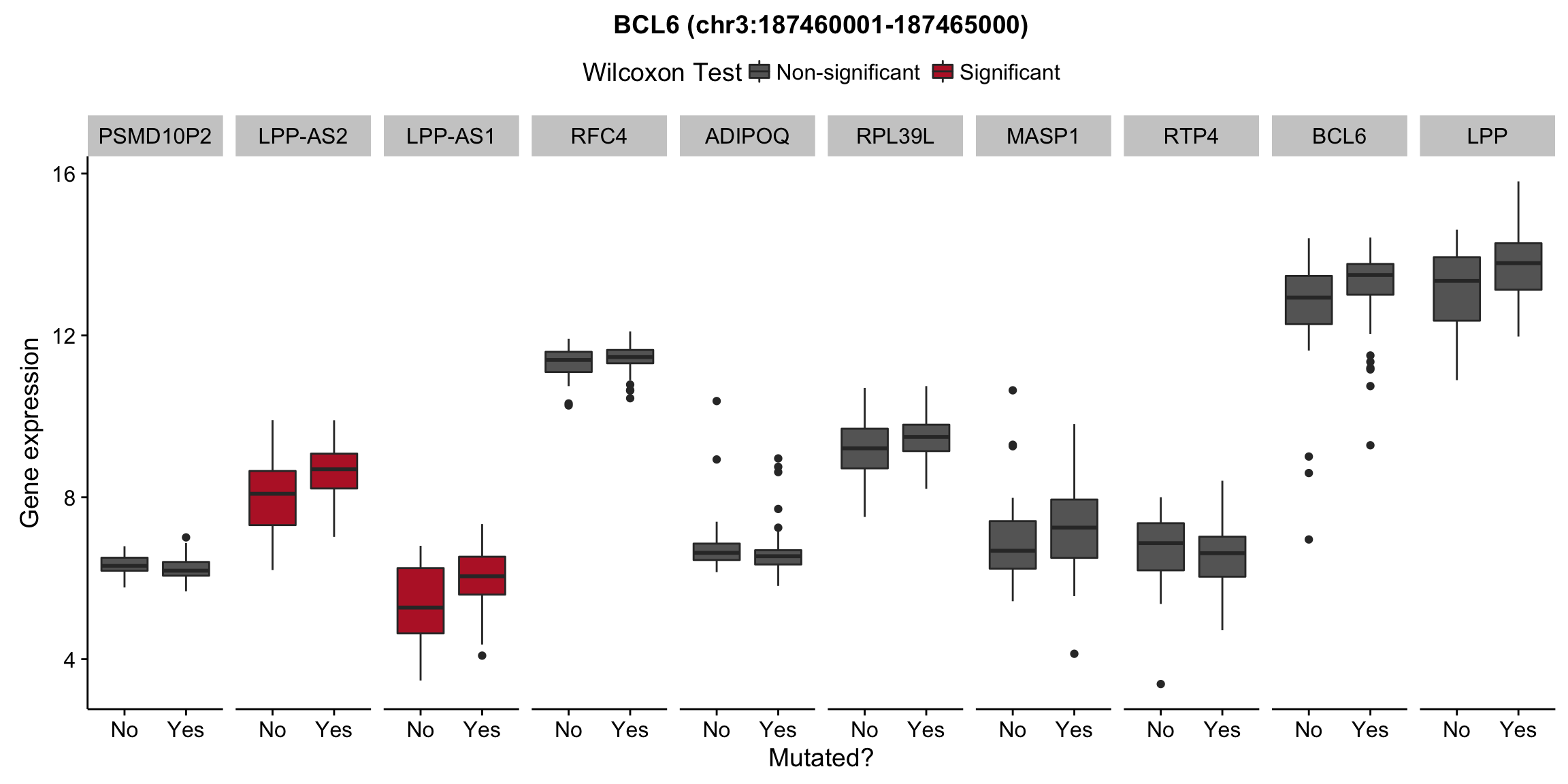
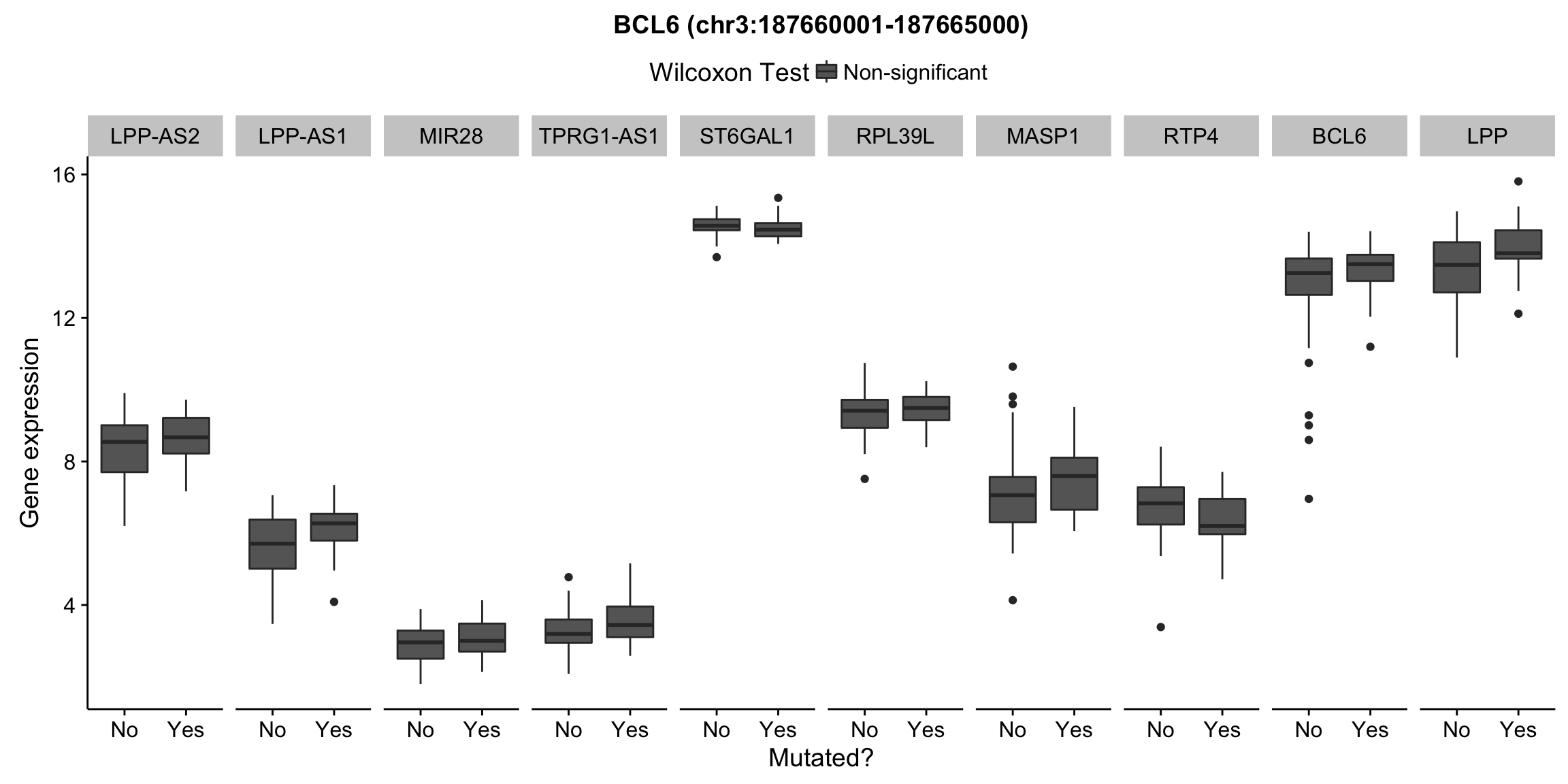
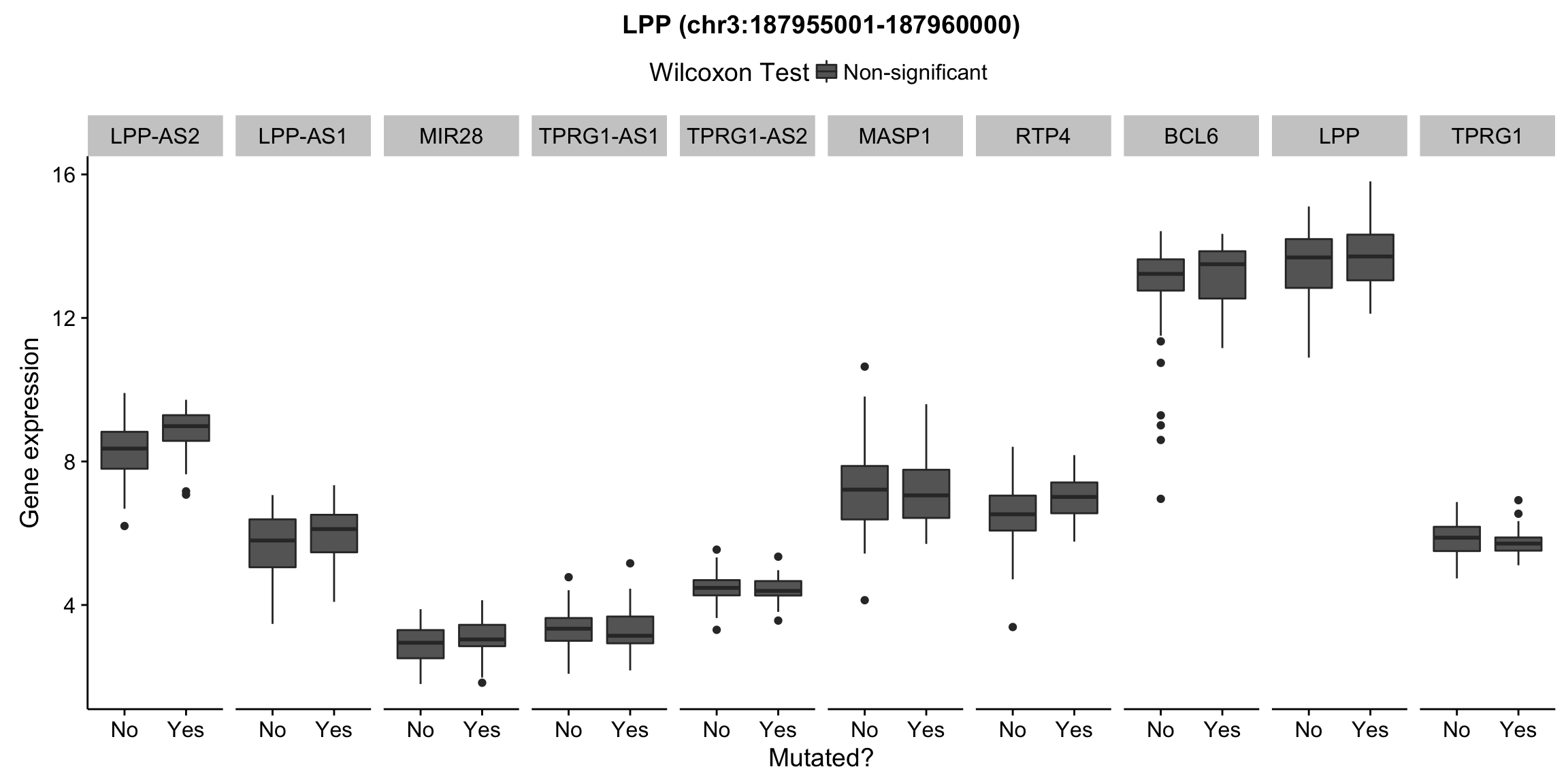
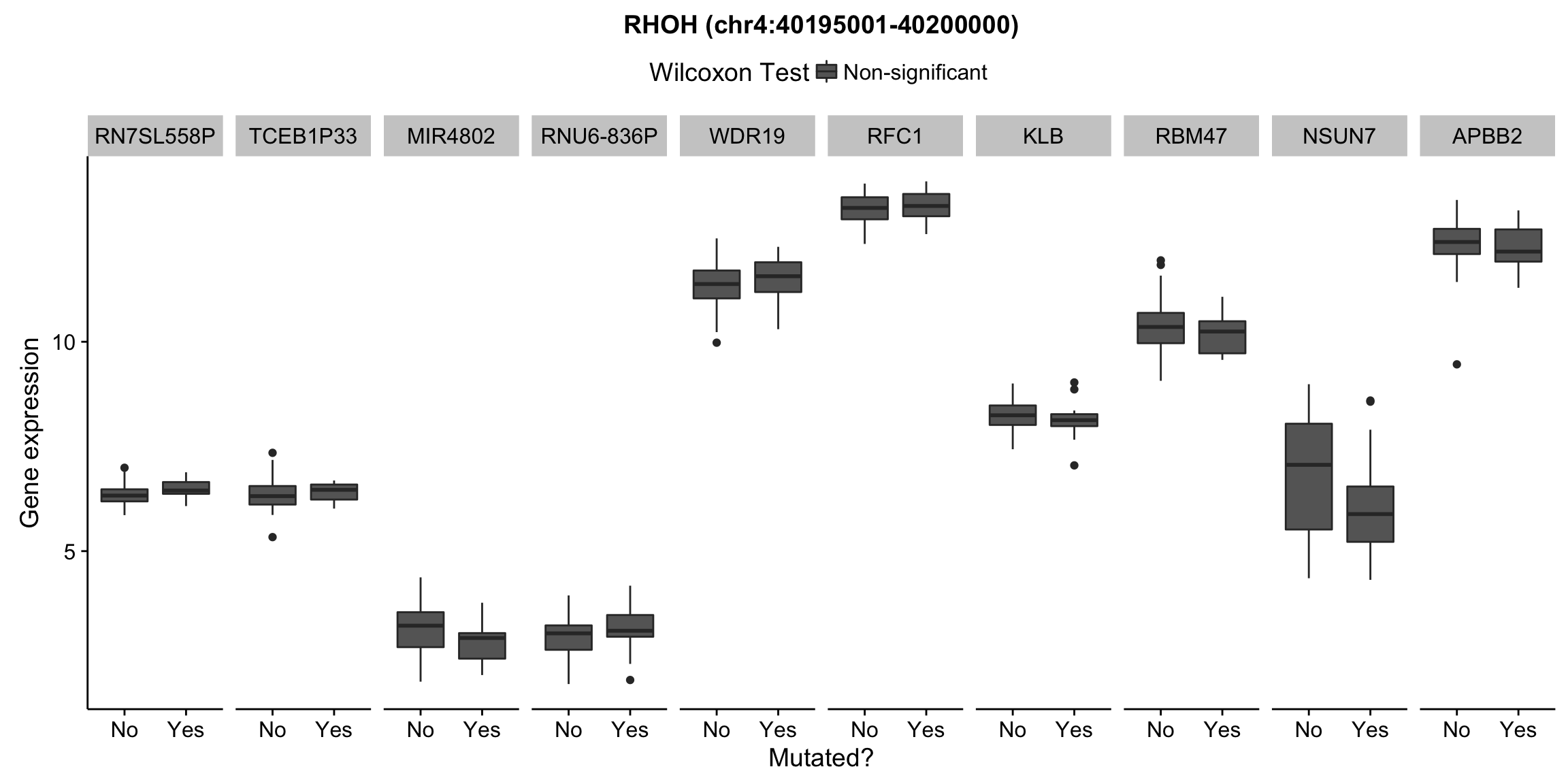
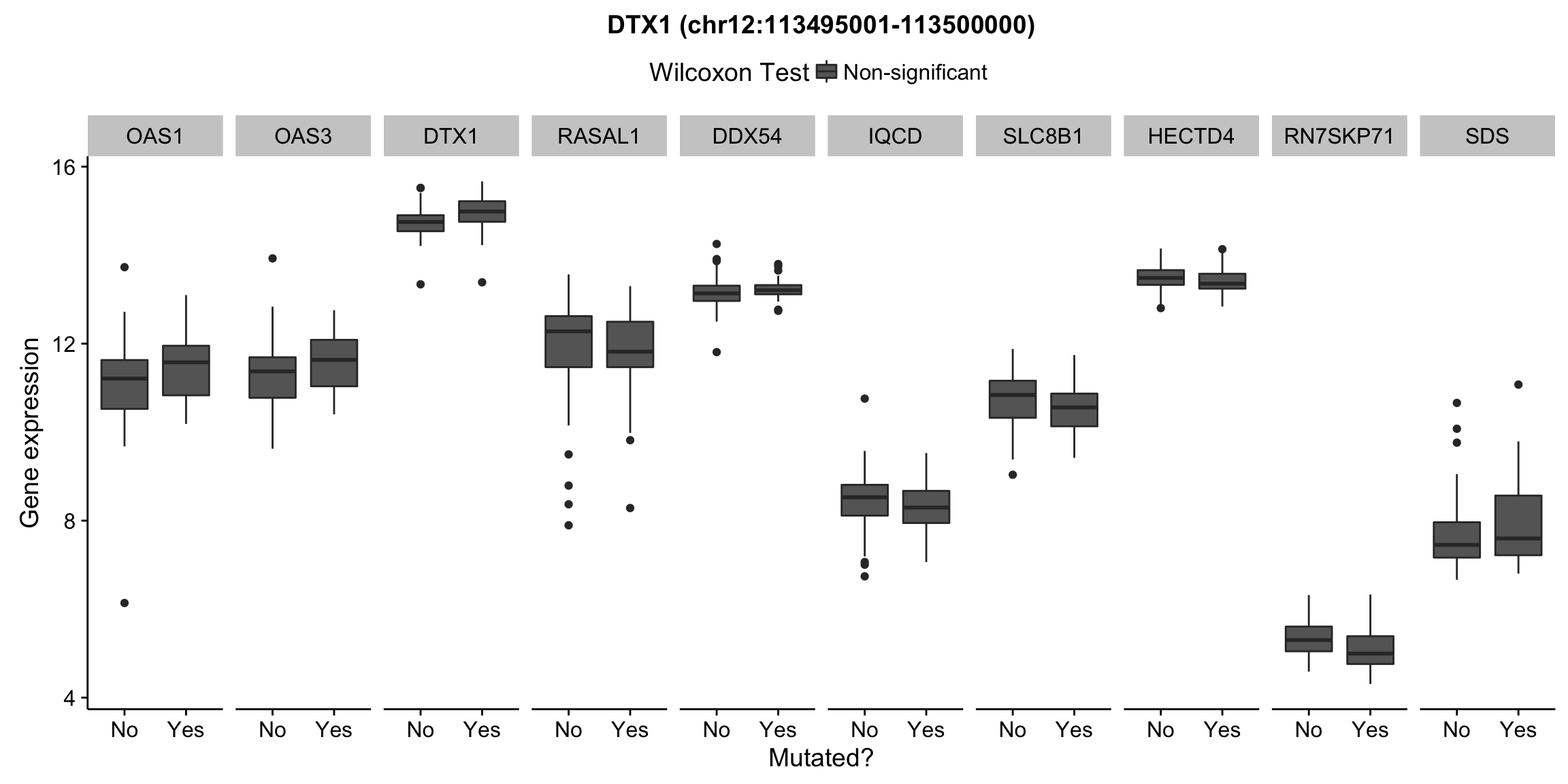
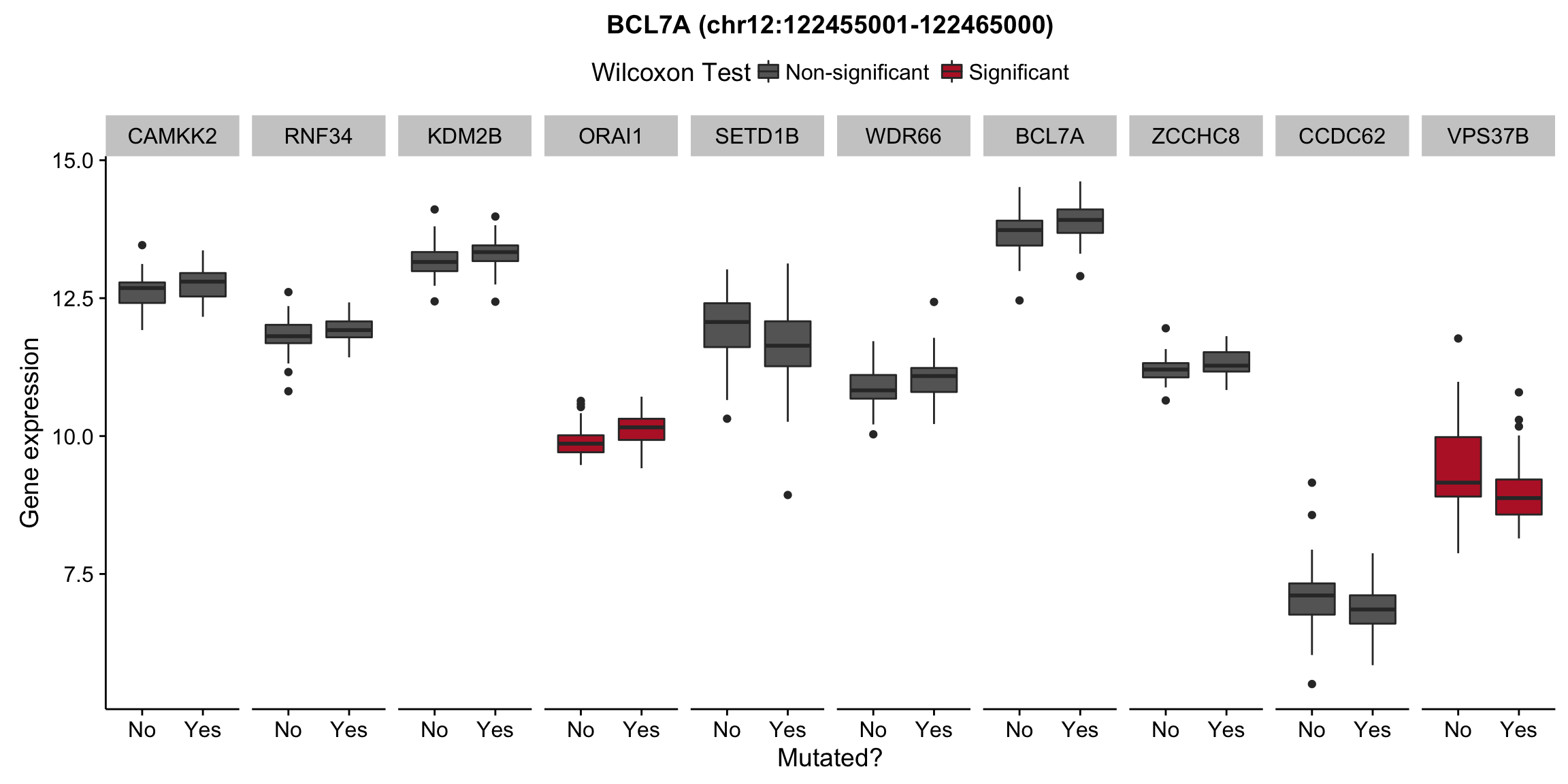
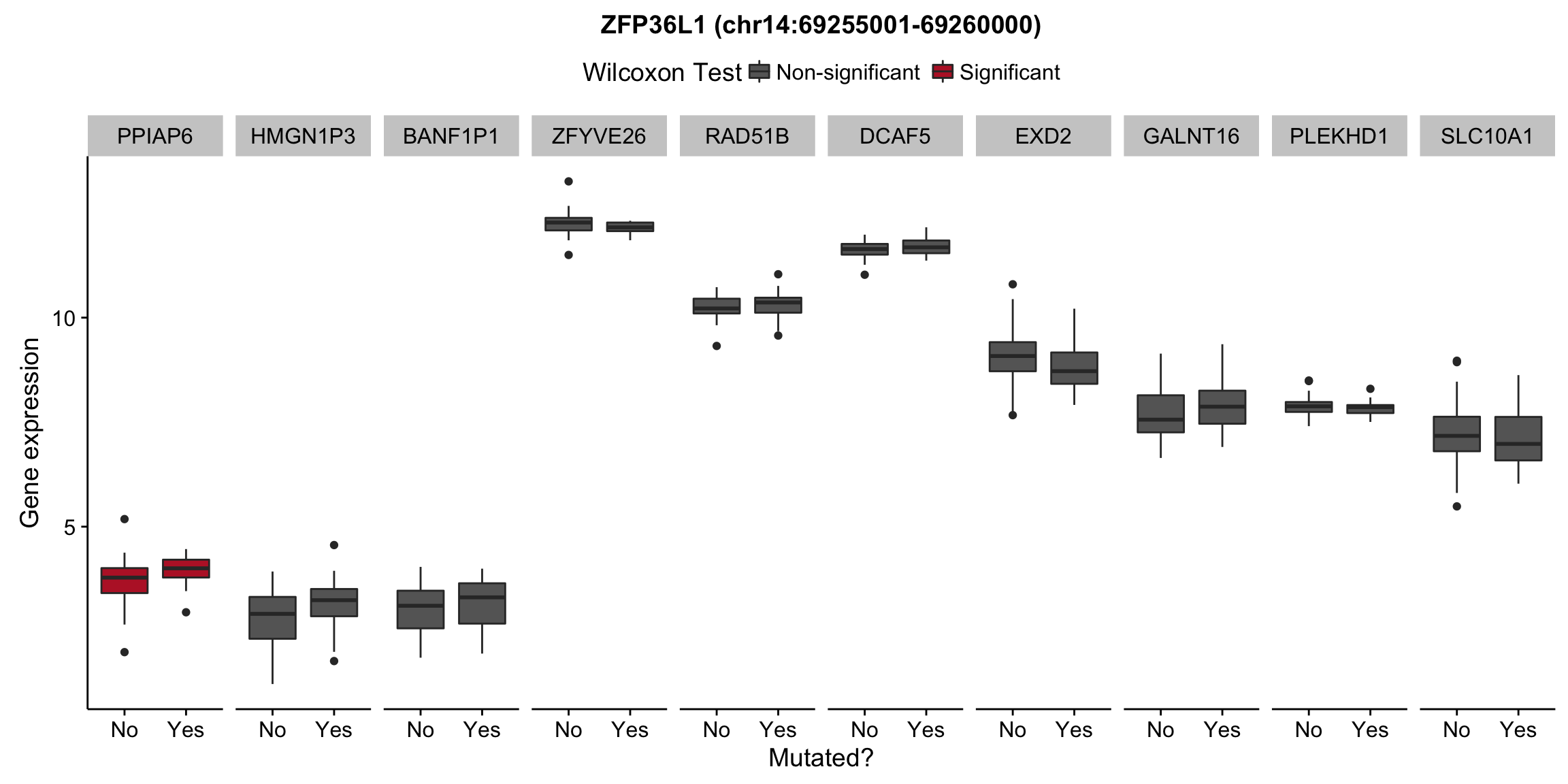
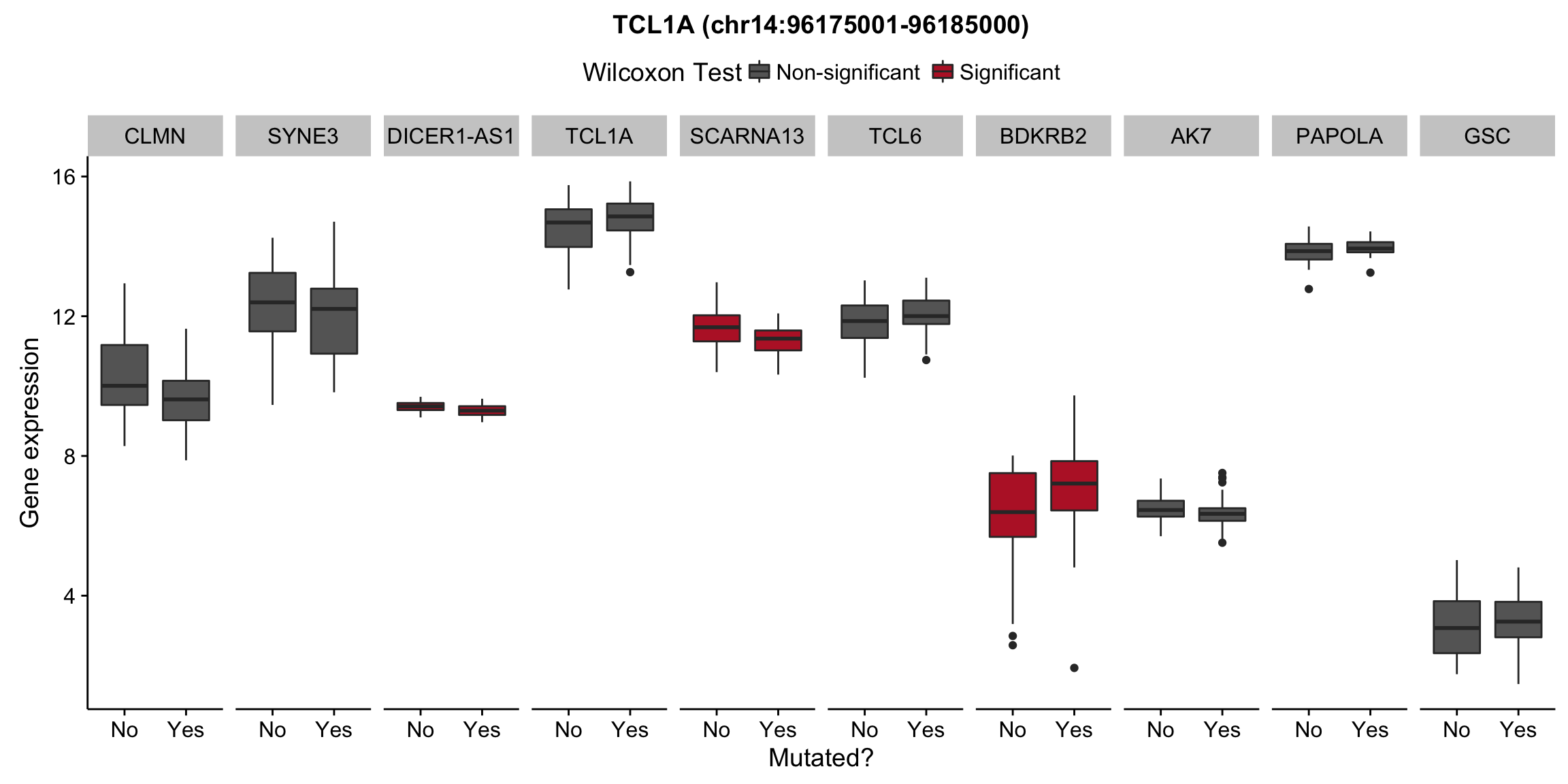
patients <- colnames(salmon$clean$counts)
mut_status <-
maf@data %>%
filter(
grepl("p[.][^=]", HGVSp_Short),
Consequence != "synonymous_variant",
Hugo_Symbol %in% smgs,
patient %in% patients) %>%
select(patient, Hugo_Symbol) %>%
distinct() %>%
mutate(Status = "Mutated") %>%
bind_rows(tibble(patient = patients, Hugo_Symbol = NA)) %>%
spread(Hugo_Symbol, Status, fill = "Unmutated") %>%
select(-`<NA>`) %>%
as.data.frame() %>%
remove_rownames() %>%
column_to_rownames("patient")
mut_colours <- list()
for (gene in names(mut_status)) mut_colours[[gene]] <- c(Unmutated = "grey75", Mutated = "grey40")
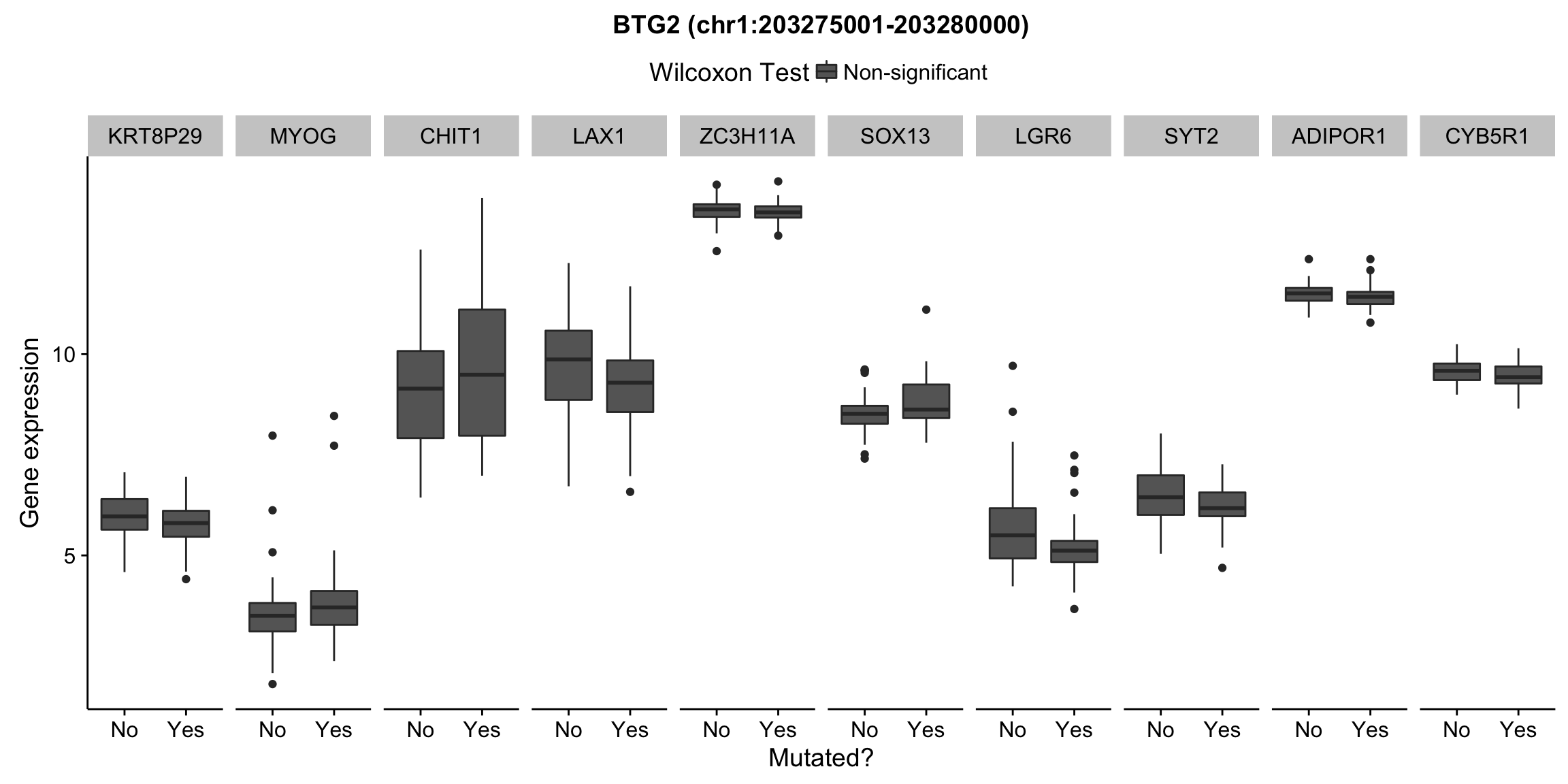
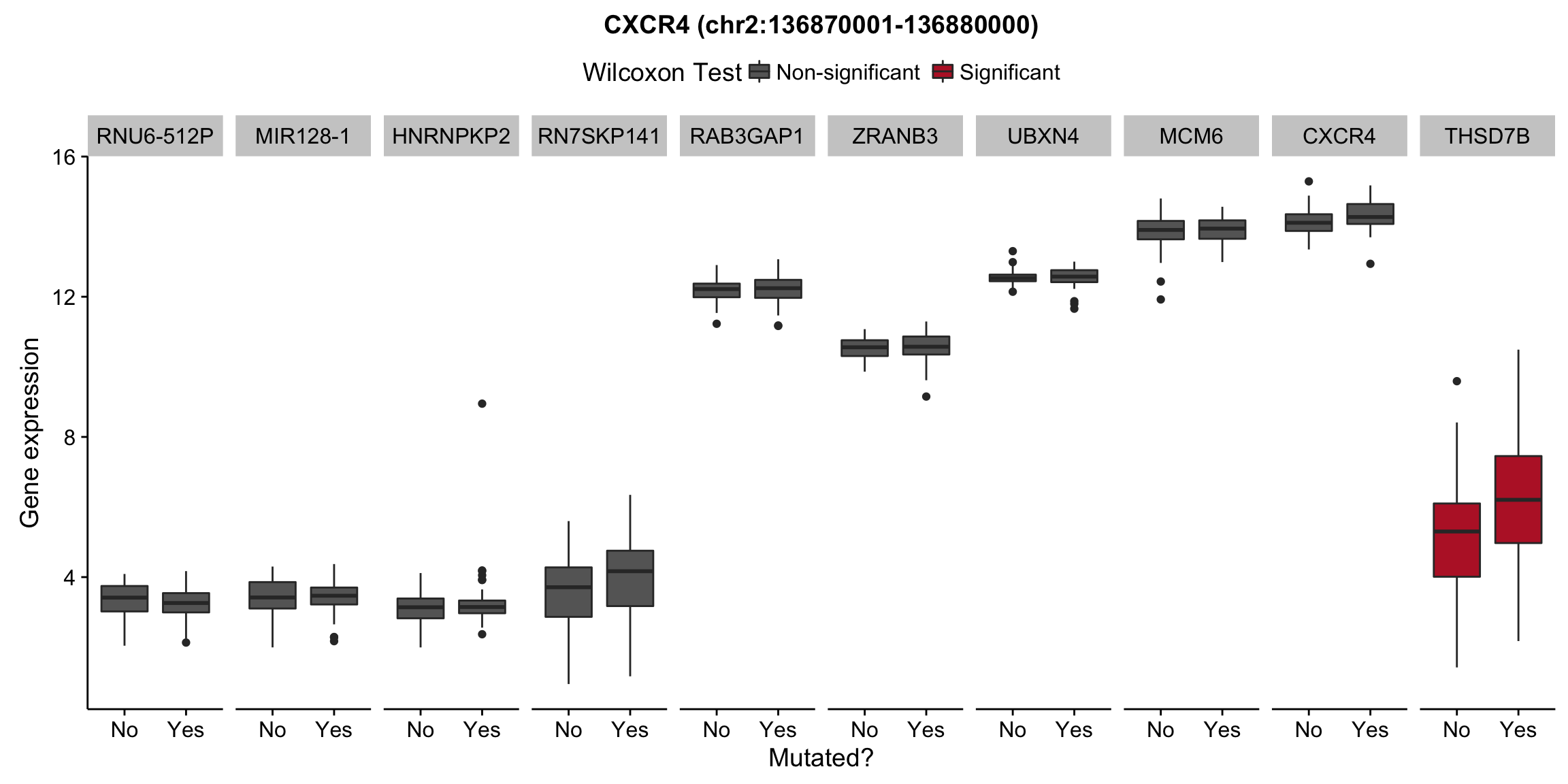
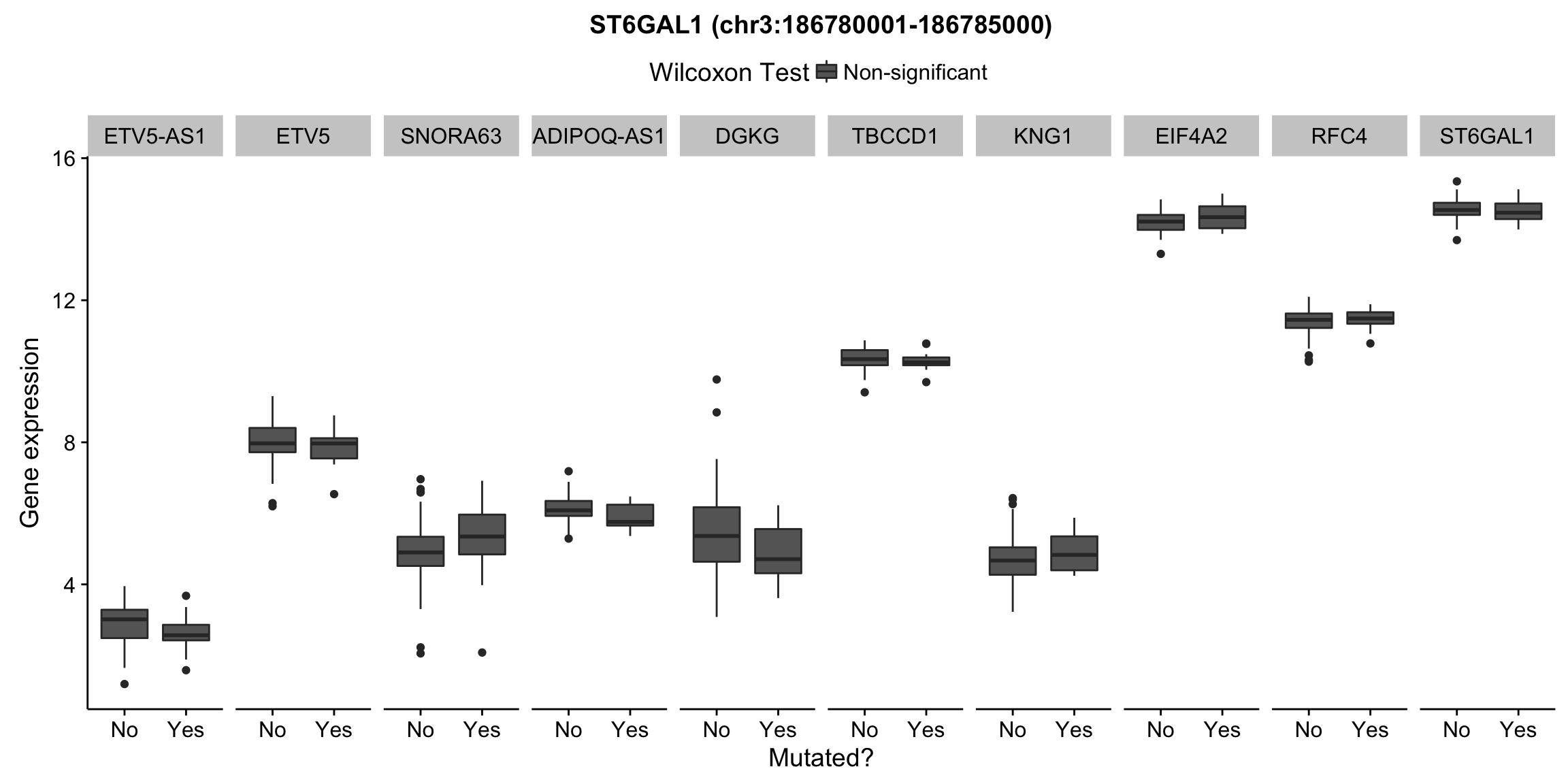
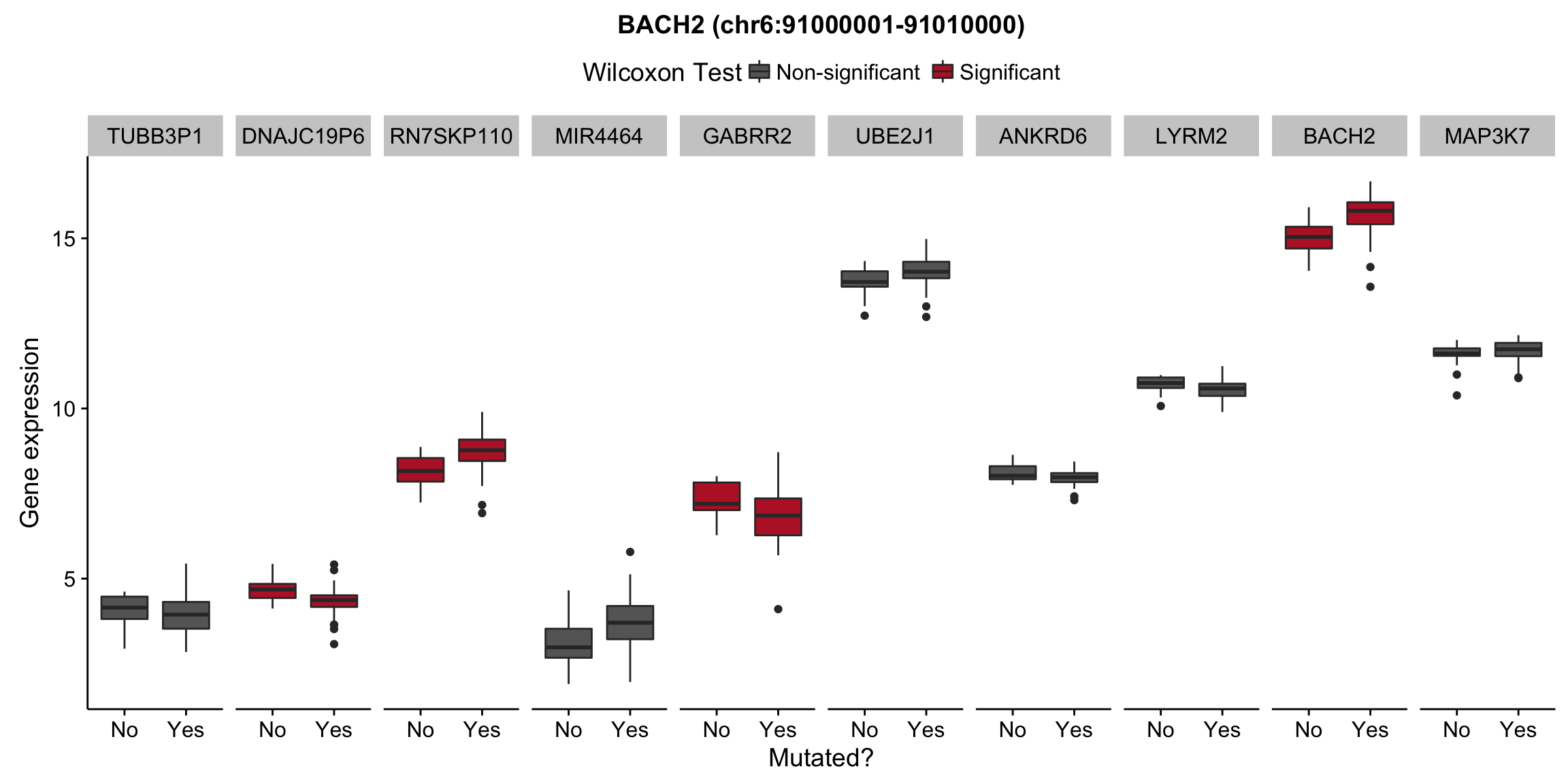
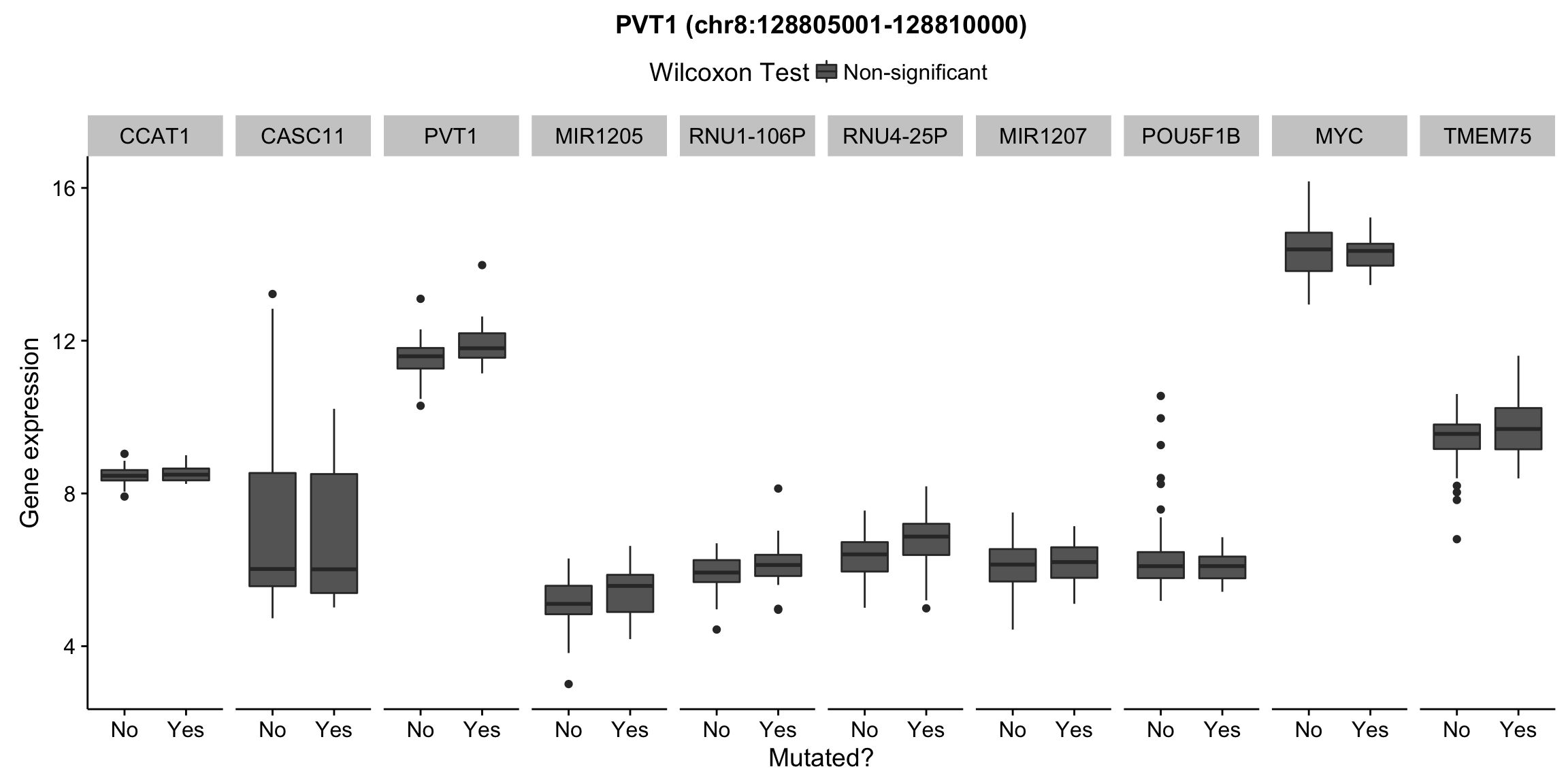
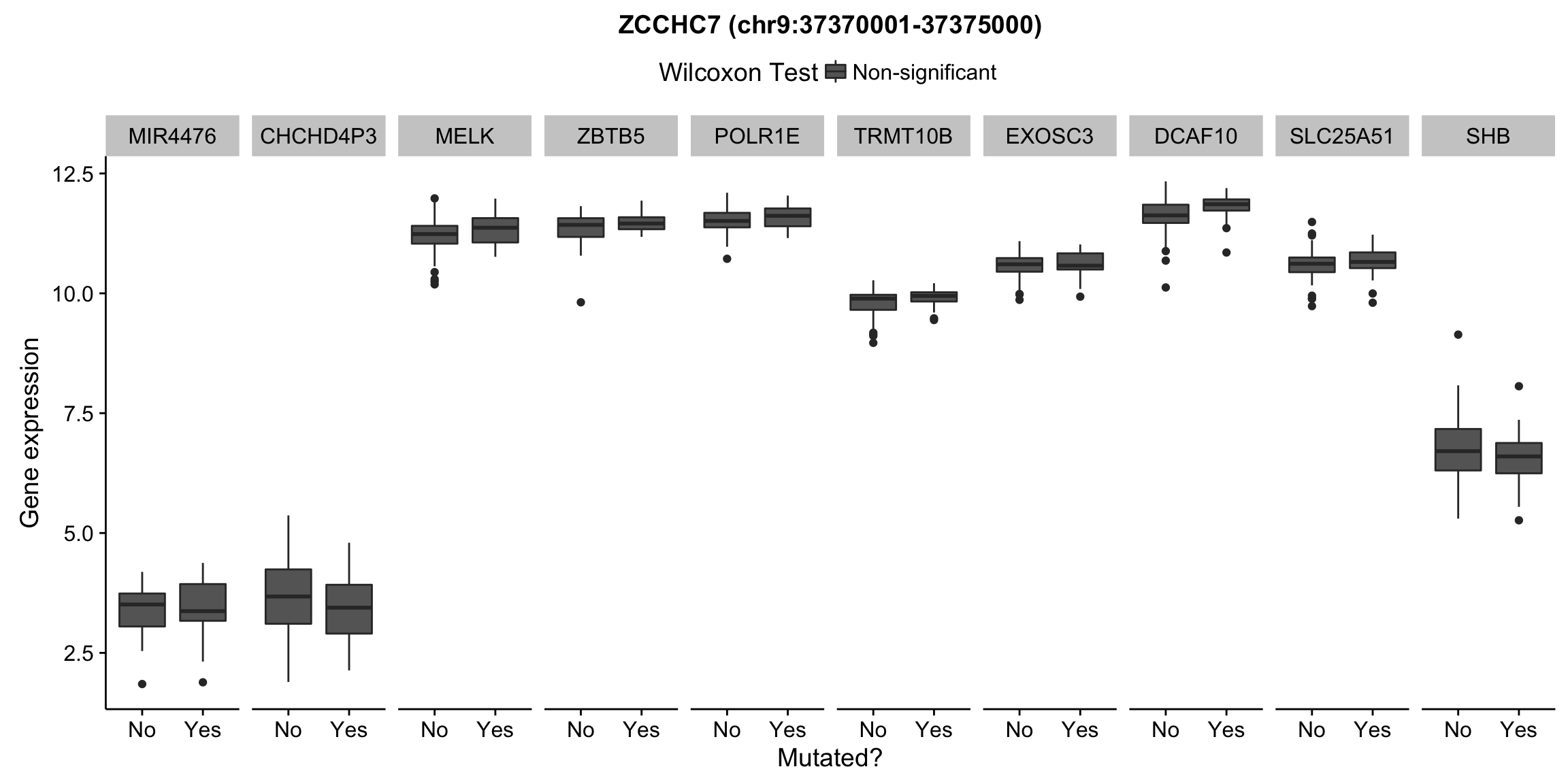
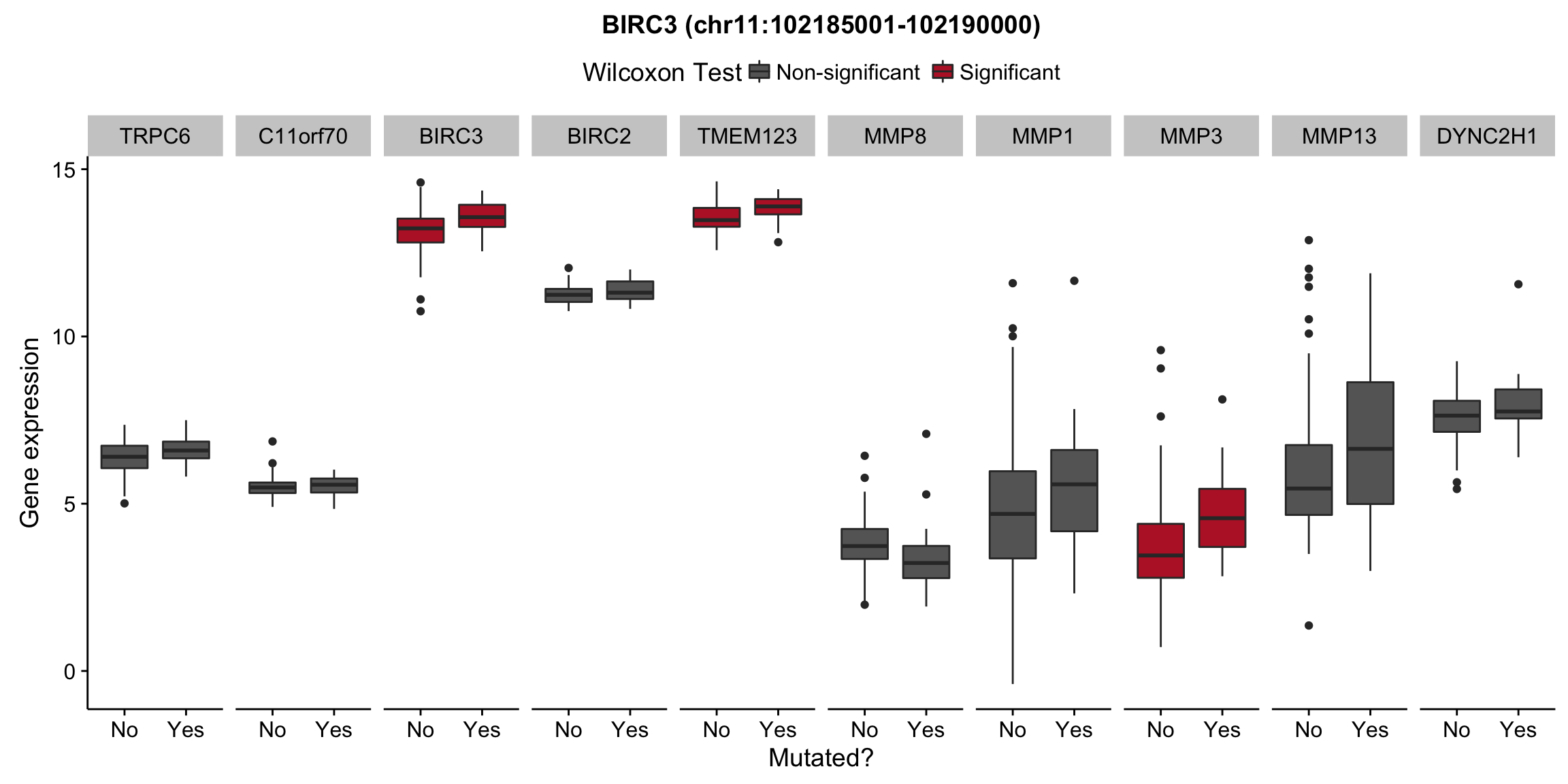
plot_nearby_gene_expr <- function(expr, genes, locus, groups, dist = 1e6, ntop = 10, title = NA) {
hits <- findOverlaps(genes, locus + dist)
nearby_genes <- genes[queryHits(hits)]$symbol
nearby_genes <- nearby_genes[nearby_genes %in% rownames(expr)]
expr_df <-
expr[nearby_genes,, drop = FALSE] %>%
t() %>%
as.data.frame() %>%
rownames_to_column("patient") %>%
bind_cols(tibble(status = groups[colnames(expr)])) %>%
drop_na() %>%
gather(gene, expr, -patient, -status) %>%
mutate(gene = fct_relevel(gene, nearby_genes)) %>%
group_by(gene) %>%
mutate(
pval = wilcox.test(expr[status], expr[!status])$p.value,
qval = p.adjust(pval, "BH"),
signif = qval < 0.01) %>%
ungroup()
top_genes <-
expr_df %>%
dplyr::select(gene, qval) %>%
distinct() %>%
top_n(10, -qval) %$%
gene
expr_df %>%
select(patient, status, gene, expr, signif) %>%
mutate(status = ifelse(status, "Yes", "No")) %>%
filter(gene %in% top_genes) %>%
ggplot(aes(x = status, y = expr, group = status, fill = signif)) +
geom_boxplot() +
facet_grid(~ gene, scales = "free_x") +
scale_fill_manual(values = c(`FALSE` = "#666666", `TRUE` = "#ba2331"),
labels = c("Non-significant", "Significant")) +
ggtitle(title) +
theme(legend.position = "top") +
labs(x = "Mutated?", y = "Gene expression", fill = "Wilcoxon Test")
}
hits <- mut_counts_df %>%
filter(signif) %>%
makeGRangesFromDataFrame(keep.extra.columns = TRUE) %>%
reduce()
plot_gene_expr_near_mutations <- function(expr, genes, hits, maf_grl,
dist = 1e6, ntop = 10) {
get_groups <- function(locus) {
map_int(as.list(maf_grl), ~countOverlaps(locus, .x)) %>%
{ . > 0 } %>% {
patient_ids <- ifelse(grepl("^BL", names(.)),
get_patient_id(names(.)),
get_icgc_donor[names(.)])
setNames(., patient_ids)}
}
get_nearest_gene <- function(x, genes_gr) {
gene_idx <- GenomicRanges::nearest(x, genes_gr)
genes_gr[gene_idx]$symbol
}
get_hit_name <- function(x, genes_gr) {
nearest_gene <- get_nearest_gene(x, genes_gr)
x <- as.data.frame(x)
paste0(nearest_gene, " (", x$seqnames, ":", x$start, "-", x$end, ")")
}
map(as.list(hits), get_groups) %>%
map2(as.list(hits), ., ~ plot_nearby_gene_expr(assay(salmon$clean$cvst), genes_gr, .x, .y,
title = get_hit_name(.x, genes_gr))) %>%
gridExtra::grid.arrange(grobs = .)
}
for (hit in as.list(hits)) {
plot_gene_expr_near_mutations(assay(salmon$clean$cvst), genes_gr, hit, maf_grl)
}