Individual
sequenza_grl <-
sequenza %>%
mutate(CNt_dis = ifelse(CNt >= 3, 1, ifelse(CNt <= 1, -1, 0))) %>%
makeGRangesListFromDataFrame("biospecimen_id", keep.extra.columns = TRUE)
seqlens <- seqlengths(BSgenome.Hsapiens.UCSC.hg38::Hsapiens)[paste0("chr", 1:22)]
bins <- tileGenome(seqlens, tilewidth = 1000000, cut.last.tile.in.chrom = TRUE)
sequenza_1mb_df <-
map(as.list(sequenza_grl), coverage, weight = "CNt_dis") %>%
map(~binnedAverage(bins, .x, "segmean")) %>%
map_df(as.data.frame, .id = "sample") %>%
select(chromosome = seqnames, start, end, segmean, sample)
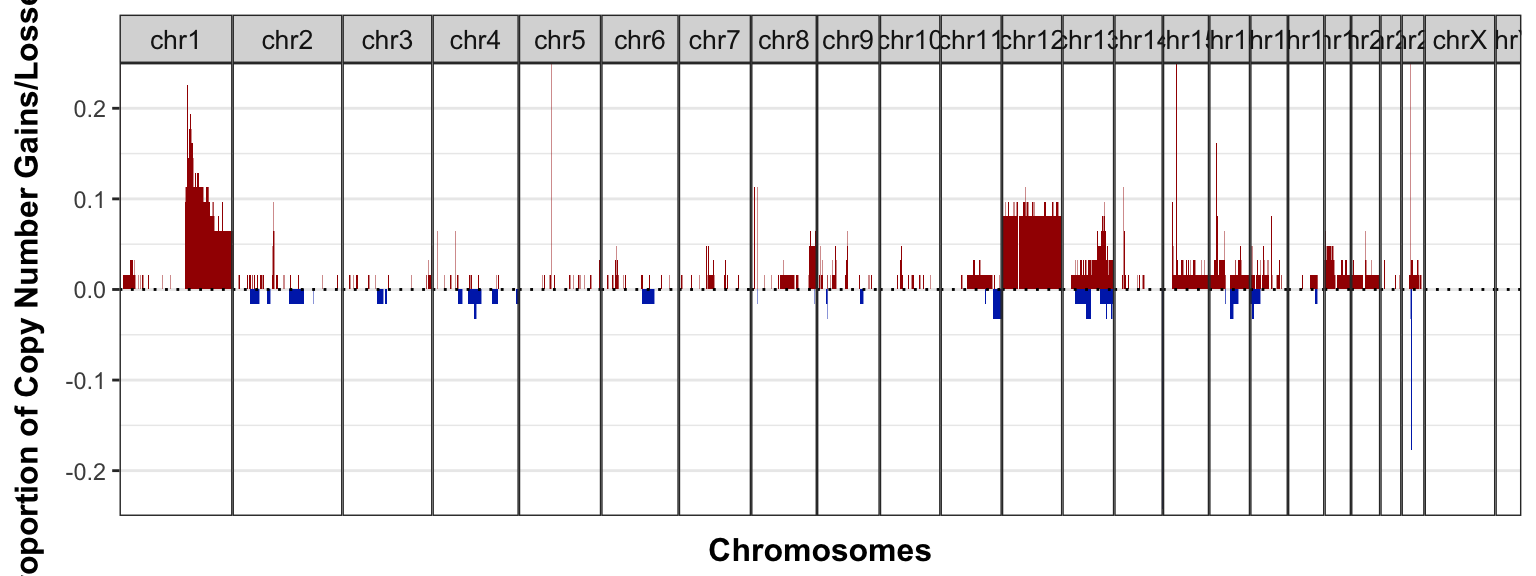
cnfreq_plot_sequenza_1mb <-
sequenza_1mb_df %>%
GenVisR::cnFreq(CN_low_cutoff = -0.5, CN_high_cutoff = 0.5,
plotChr = paste0("chr", c(1:22, "X", "Y")), genome = "hg38") +
theme(panel.spacing = unit(0, "lines")) +
coord_cartesian(ylim = c(-0.25, 0.25))
cnfreq_plot_sequenza_1mb
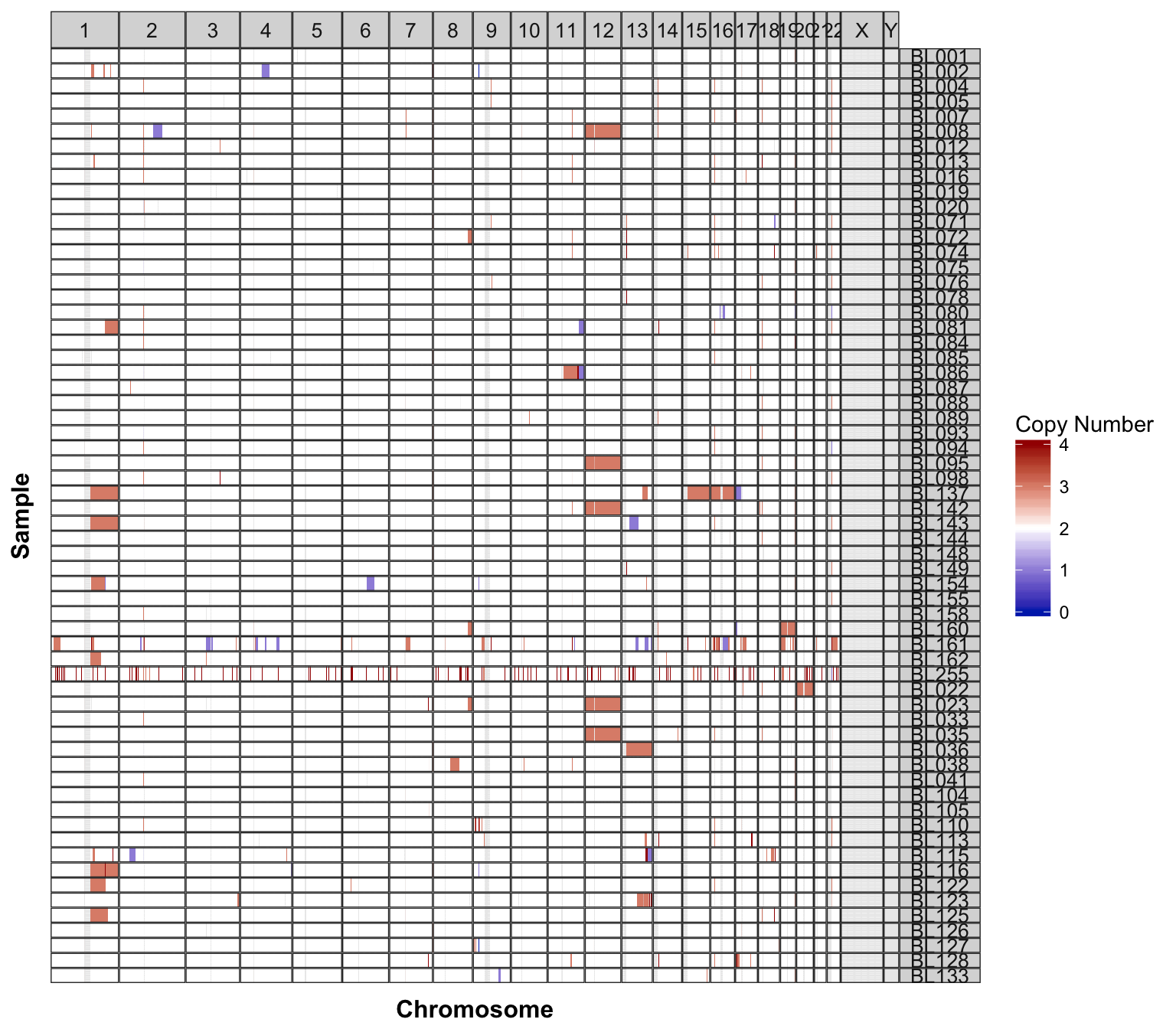
cnspec_plot_sequenza <-
sequenza %>%
select(chromosome, start, end, segmean = CNt, sample = patient) %>%
GenVisR::cnSpec(genome = "hg38", ) +
theme(panel.spacing = unit(0, "lines"))
cnspec_plot_sequenza
sequenza_gistic = readGistic(
gisticAllLesionsFile = file.path(paths$gistic, "all_lesions.conf_90.txt"),
gisticAmpGenesFile = file.path(paths$gistic, "amp_genes.conf_90.txt"),
gisticDelGenesFile = file.path(paths$gistic, "del_genes.conf_90.txt"))
plotGisticResults(sequenza_gistic)
maf_gistic = read_maf(
paths$maf,
gisticAllLesionsFile = file.path(paths$gistic, "all_lesions.conf_90.txt"),
gisticAmpGenesFile = file.path(paths$gistic, "amp_genes.conf_90.txt"),
gisticDelGenesFile = file.path(paths$gistic, "del_genes.conf_90.txt"))
oncoplot(maf_gistic, genes = smgs, removeNonMutated = FALSE)