
|
|
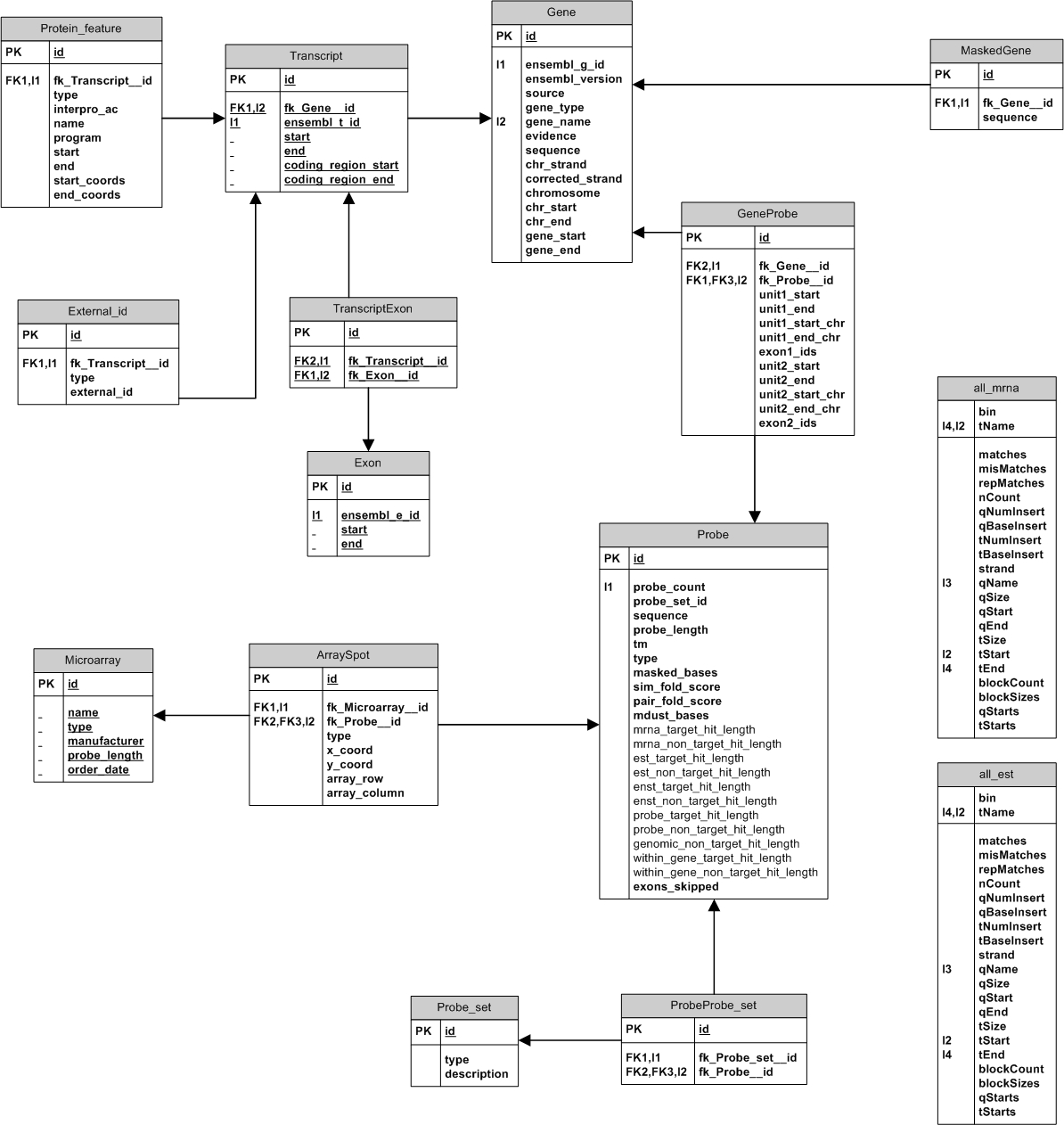
Array Design Method Following is a brief description of the process by which the ALEXA platform is used to create a custom array design. For a more detailed description of how to use ALEXA, refer to the user manual or the platform code itself. Database population A single database instance is created for each species and initially populated with information from EnsEMBL (using the EnsEMBL API). This database will store all probe sequences and the results of all quality tests for these probes. Each probe will also be annotated by creating associations with genes, transcripts, exons, protein motifs, transmembrane domains, etc. For each probe, coordinates are provided to describe the position of the probe relative to the gene sequence as well as the chromosome it resides on. Refer to the database schema for more details. Once the database is populated with gene models, basic statistics are generated to describe the number of transcripts per gene, the size of transcripts and exons, the number of known exon-skipping events, etc. Refer to the pre-computed designs section to see examples of this. Repeat Masking To help avoid probes that correspond to repetitive sequences the complete gene sequence of every imported gene is repeat masked with updated repeat libraries from the Genetic Information Research Institute. A probe not allowed if more than 25% of its sequence is masked. Probe Sequence Extraction Probes are extracted for all known, non-pseudo EnsEMBL genes. Five probe types are generated: Exon, Intron,
Exon-Junction, Exon-Boundary and Random Negative control (See Intro section for description of these).
Probe extraction can also be specified for predicted genes and this is recommended for genomes with preliminary levels of
annotation where most genes are still considered to be predicted. The user can also specify to use an isothermal or fixed-length design.
In an isothermal design, the length of each probe is allowed to vary to achieve a specified target Tm. This approach
is recommended for users who will have their arrays manufactured by NimbleGen Systems Inc. Balancing the Tm of
probes across the array potentially reduces the variable performance of probes with dramatically different GC content
which are all hybridized at a single hybridization stringency. As probes are extracted the Tm of each probe is calculated
by a nearest neighbor method and simple tests are conducted to avoid ambiguous bases and repetitive elements. Probe Quality Testing All probe sequences are subjected to the following quality tests in an effort to identify unsuitable probes: Probe Filtering Using the quality metrics described in the previous steps a filtered probe set is selected from the complete probe population. Probes which fail the quality tests above are excluded at this time. At this time the 'best' n probes are selected for each exon or intron (where n is the desired probeset size). In the pre-computed designs 3 probes are selected for each probeset. Summarizing and Visualizing an ALEXA Design Once the filtering and selection of probes for each gene is complete, the entire design is summarized. Various statistics are generated describing the thermodynamic properties and specificity of the probes as well as the success of probe design for each gene (probe coverage %), etc. An annotation file summarizing the features of each gene such as the number of transcripts, exons, probes selected, etc. is also generated and provides links to custom UCSC tracks which display the genomic position of all probes for a particular gene. Design Validation and Submission A brief manual validation of ALEXA designs can be accomplished by visualizing probe coordinates and sequence
in the custom UCSC tracks described above for a small number of genes of interest.
Once the comfortable with the design the user generates a submission file and sends it to an array synthesis facility.
This can be done in-house in the case of spotted oligo arrays or by a custom array service such as those provided by NimbleGen. |
{kind=link}