|
|
Introduction In metazoan genomes, the process of gene expression involves production of a messenger RNA (mRNA) molecule by transcript initiation, splicing and poly-adenylation. For many genes, multiple mRNA isoforms can be produced by alternative transcript initation, alternative splicing and alternative poly-adenylation. We refer to these processes collectively as alternative expression or 'AE'. Alternative expression can result in alternative mRNA isoforms with subtle or dramatic differences in exon sequence content. We previously reviewed bioinformatic, microarray and sequence based methods for analysing alternative expression (see Griffith and Marra, 2007). This webpage provides supplementary materials for a publication describing the use of 'next generation' massively parallel RNA sequencing to identify and quantify alternative mRNA isoforms. Alternative expression analysis by massively parallel RNA sequencing Massively parallel RNA sequencing involves the random sequencing of an RNA sample by generation of large numbers (10s to 100s of millions) or short reads (25-300bp depending on the platform). The majority of massively parallel sequencing conducted to date has involved one of three sequencing platforms offered by Illumina (Solexa), ABI (SOLiD) and Roche (454). Recent reports have described the use of these platforms to perform gene expression analysis and in a few cases identify and quantify specific mRNA isoforms. The following report in principle, the use of massively parallel sequencing for transcriptome analysis: Cloonan et al. 2008, Li et al. 2008, Pan et al. 2008, Sultan et al. 2008, Tang et al. 2009, and Wang et al. 2008. In addition to these, proof-of-principle reports, several tools designed specifically to utilize massively parallel RNA sequencing for some aspect of alternative expression
analysis have been released. These tools are generally focused on either gapped alignment of short reads or de novo assembly and characterization of transcript models.
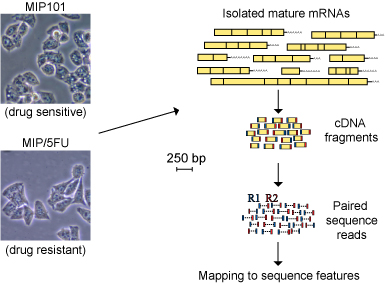
Recently released tools in this area include: Contribution and novelty While the preliminary reports and tools listed above showed the promise of next-generation sequencing for transcriptome analysis, we developed novel methods for assessing the expression, differential expression and alternative expression of known and predicted mRNA isoforms including metrics for identifying reciprocal expression of alternative isoforms. We also developed a novel method for assessing the expression of sequence features above background while taking into account locus-by-locus noise levels. We make available databases of sequence features and an associated visualization tool specifically tailored to the analysis of alternative isoforms by WTSS, including information on the sequence support, conservation and protein coding effect of each feature. These feature databases may be immediately useful for expression analysis of specific isoforms. For example, when multiple transcripts are known for a single locus, the exons (or portions of exons in the case of overlaping exons with alternative boundaries) and junctions that distinguish each known transcript are provided. Similarly we provide annotations for thousands of known and predicted exon skipping events, putative cryptic exons, intron retentions, and so on. Finally, we report the first application of WTSS to a model of chemotherapy resistance in colorectal cancer. While the data we used to validate our method relates to this specific area of cancer biology research, we believe that this work represents a general model for comparison of relevant disease states such as cancer and normal tissue, pre- and post-treatment biopsies, etc. With this website we make available: sequence data, alternative expression annotation databases (for human, chimp, mouse, rat, chicken fruit fly, and yeast), candidate gene lists, source code, visualizations and other resources to facilitate alternative expression analysis by massively parallel RNA sequencing. Refer to Griffith et al. for additional details. Experimental overview (validation experiment) Total RNA was isolated from samples to be compared. In this example, 5-FU sensitive and resistant colorectal cancer cell lines. Messenger RNA is purified by polyA+ selection, followed by cDNA generation with random hexamers, fragmentation by sonication, selection of cDNA fragments by gel electrophoresis, ligation of sequencing linkers and sequencing of the ends 100’s of millions of such fragments with an Illumina GAII sequencing device. The resulting are then mapped to a database of sequence features defined by compilation of genome, transcriptome and expressed sequence databases. Refer to Griffith et al. for additional details.
|