https://github.com/bcgsc/xmatchview
XMatchView is written in python and runs on linux and windows. It serves as a visual tool for analyzing cross_match alignments. Cross_match (Green, P. (1994) http://www.phrap.org) uses an implementation of the Smith-Waterman algorithm for comparing DNA sequences that is sensitive.
Examples
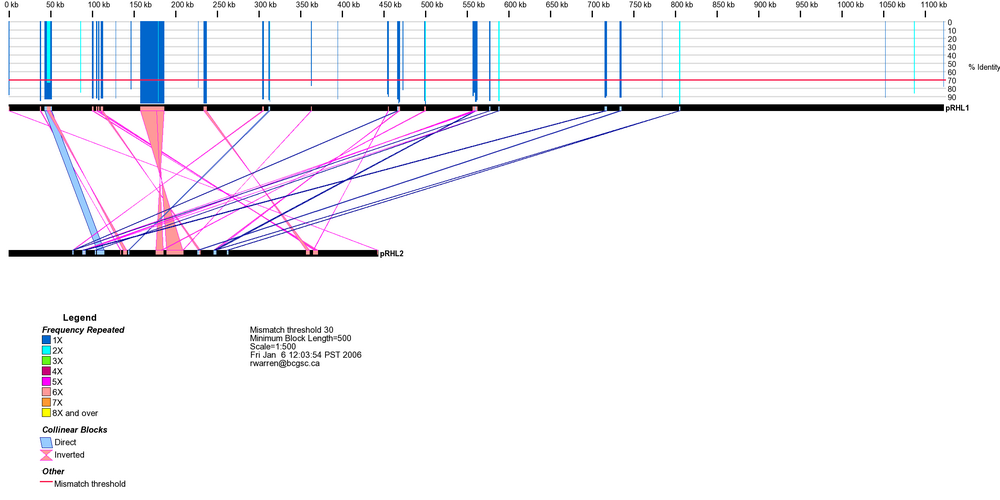
DNA sequence alignment between Rhodococcus sp. RHA1 plasmid pRHL1 and pRHL2 shows a 28 kb duplicated segment sharing more than 99% sequence identity between the two linear plasmids. A 4 kb insertion in the corresponding plasmid 2 segment indicates that pRHL1 likely contains the ancestral copy.

A fully finished chromosome from Cryptococcus gattii strain WM276 is aligned to a contig from the C.neoformans strain B3501 whole-genome shotgun assembly. There are high levels of conservation and obvious rearrangements between the two human pathogens. Differences in the alignment may enable the discovery of virulence genes and help retrace the evolution history between these sister strains.
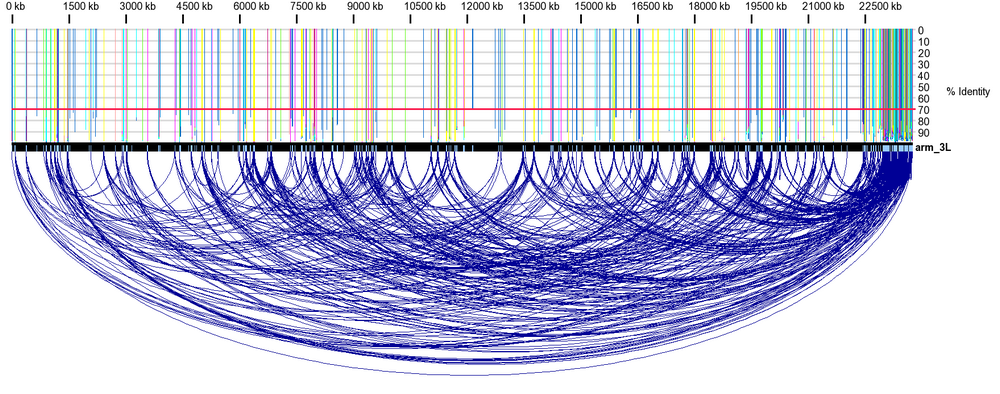
DNA repeats within Drosophila melanogaster arm 3L. Notice the repeat density and frequency near the telomere, on the 3' end of the chromosome arm.
About the graphs
DNA sequences are represented by the black rectangles. Direct and inverted repeats are depicted by blue- and salmon-colored boxes on the DNA sequence, respectively. Only collinear blocks having less than user-defined % base mismatch are displayed, along with their relationship. A sliding window is used to determine the frequency of repeated bases between or within DNA sequences. The frequency is depicted by the blue, green, red and yellow lines above the top DNA sequence. The length of each line represents the percent sequence identity shared by the two sequences inside that window.
Main Features
XMatchView allows users to:
- Easily identify collinear blocks
- Assess the relationship between collinear blocks
- Analyze the sequence identity between repeated segments
- View the repeat frequency
- Launch cross_match directly from a GUI
- Produce publication-quality graphs in a variety of image formats including jpeg, png, gif, tiff, bmp and ps
License
Copyright (c) 2006-2018 Canada's Michael Smith Genome Science Centre. All rights reserved.
This program is free software; you can redistribute it and/or modify it under the terms of the GNU General Public License as published by the Free Software Foundation; either version 2 of the License, or (at your option) any later version.
This program is distributed in the hope that it will be useful, but WITHOUT ANY WARRANTY; without even the implied warranty of MERCHANTABILITY or FITNESS FOR A PARTICULAR PURPOSE. See the GNU General Public License for more details.
Documentation
Refer to the README file in the download to learn about the program requirements and how to install.
Credits
Rene Warren
Funding for this project was provided by Genome BC and Genome Canada.
